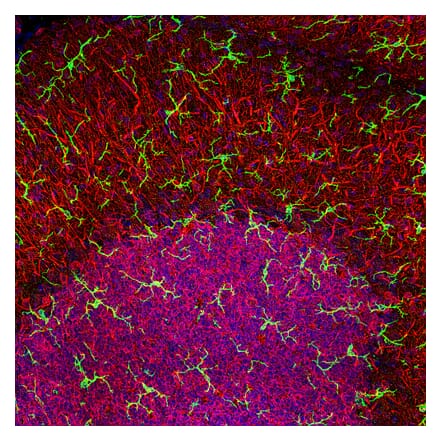
Anti-Iba1 Antibody (A82670) | |
100µg/$4951 Citation | |
| Description: | Goat polyclonal antibody to Iba1. |
| Applications: | ELISA, WB, IHC |
| Reactivity: | Human, Mouse, Rat |
| Conjugate: | Unconjugated |
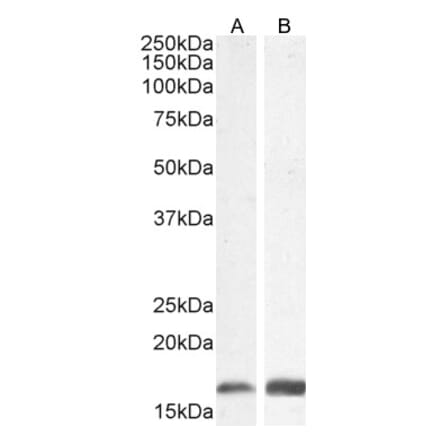
Anti-GFAP Antibody (A85419) | |
10µl - 100µl/$190 – $5451 Review4 Citations | |
| Description: | Rabbit polyclonal antibody to GFAP. |
| Applications: | WB, ICC/IF, IHC |
| Reactivity: | Human, Horse, Bovine, Porcine, Rat, Mouse |
| Conjugate: | Unconjugated |
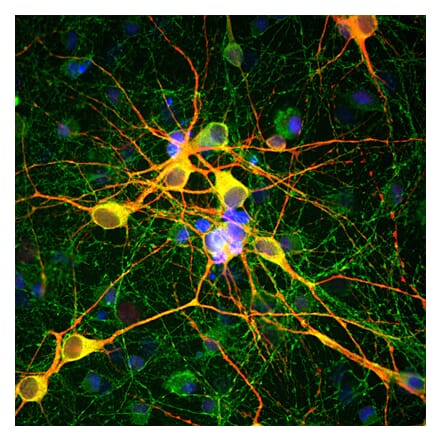
Anti-MAP2 Antibody (A85363) | |
5µl - 50µl/$190 – $545 | |
| Description: | Chicken polyclonal antibody to MAP2. |
| Applications: | WB, ICC/IF, IHC |
| Reactivity: | Human, Rat, Mouse |
| Conjugate: | Unconjugated |
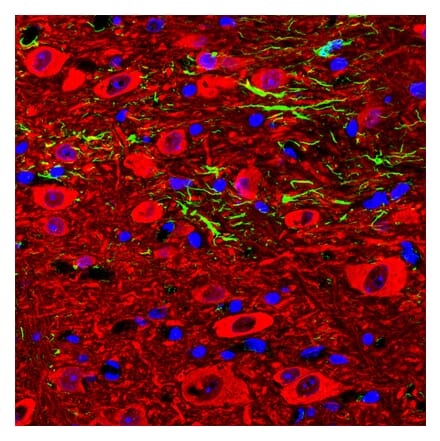
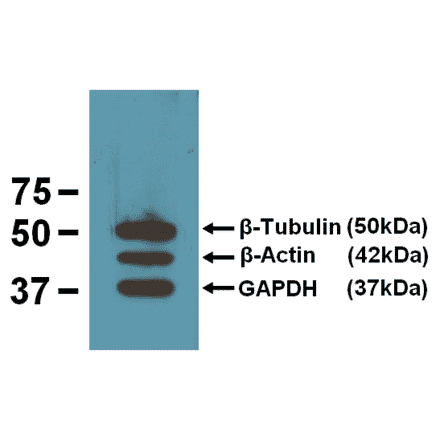
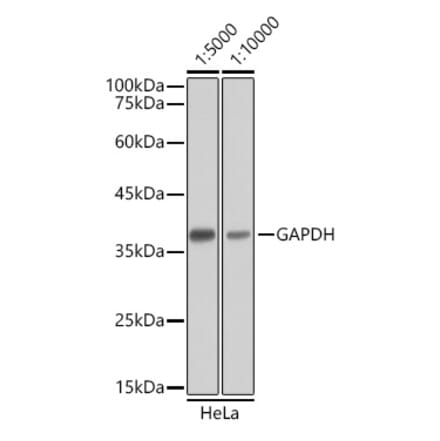
Anti-GAPDH Antibody [1D4] (A85382) | |
10µl - 100µl/$190 – $5451 Citation | |
| Description: | Mouse monoclonal [1D4] antibody to GAPDH. |
| Applications: | WB, ICC/IF, IHC |
| Reactivity: | Human, Rat, Mouse, Bovine, Porcine, Horse, Monkey, Canine, Chicken |
| Conjugate: | Unconjugated |
![Immunofluorescence - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_1.jpg?profile=product_search)
Anti-GFAP Antibody (A85307) | |
10µl - 100µl/$190 – $545 | |
| Description: | Chicken polyclonal antibody to GFAP. |
| Applications: | WB, ICC/IF, IHC |
| Reactivity: | Human, Horse, Cow, Porcine, Rat, Mouse |
| Conjugate: | Unconjugated |
Anti-PCNA Antibody [PC10] (A86878) | |
10µg - 100µg/$200 – $340 | |
| Description: | Mouse monoclonal [PC10] antibody to PCNA. |
| Applications: | Flow Cytometry, IP, WB, IHC-P, IHC-Fr, ICC |
| Reactivity: | Mouse, Human, Non-Human Primates, Drosophila, Chicken, Rat |
| Conjugate: | Unconjugated |
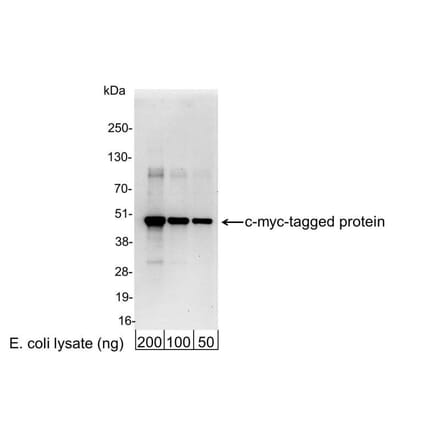
Anti-Myc Tag Antibody [9E10] (A85464) | |
10µg - 100µg/$200 – $365 | |
| Description: | Mouse monoclonal [9E10] antibody to Myc Tag. |
| Applications: | Flow Cytometry, IP, WB, IHC-P, Flow Cytometry (Intracellular) |
| Reactivity: | Human, Epitope Tag |
| Conjugate: | Unconjugated |
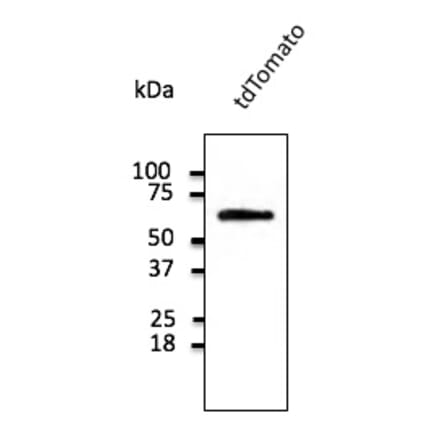
Anti-tdTomato Antibody (A121690) | |
600µg/$55522 Citations | |
| Description: | Goat polyclonal antibody to tdTomato. |
| Applications: | WB, IF, IHC-P, IHC-Fr, IEM |
| Reactivity: | tdTomato, mCherry, RFP |
| Conjugate: | Unconjugated |
Anti-His Tag Antibody [HIS.H8] (A85277) | |
10µg - 200µg/$200 – $5651 Citation | |
| Description: | Mouse monoclonal (HIS.H8) antibody to His Tag. |
| Applications: | Dot, ELISA, IP, IS, WB |
| Isotype: | IgG2b |
| Conjugate: | Unconjugated |
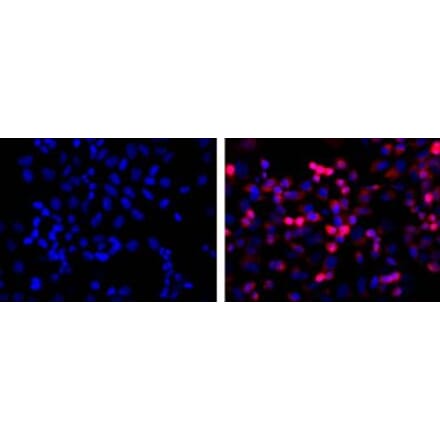
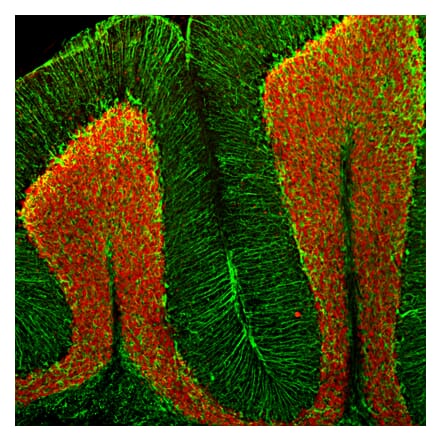
Anti-Fibrillarin Antibody [38F3] (A85370) | |
50µl - 500µl/$190 – $5451 Citation | |
| Description: | Mouse monoclonal [38F3] antibody to Fibrillarin. |
| Applications: | WB, ICC/IF, IHC, Flow Cytometry |
| Reactivity: | Human, Rat, Mouse, Drosophila, C. elegans, Saccharomyces cerevisiae |
| Conjugate: | Unconjugated |
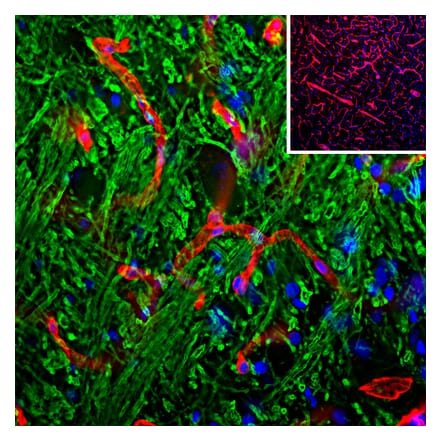
Anti-NeuN Antibody [1B7] (A85405) | |
10µl - 100µl/$190 – $5451 Citation | |
| Description: | Mouse monoclonal [1B7] antibody to NeuN. |
| Applications: | WB, ICC/IF, IHC |
| Reactivity: | Human, Rat, Mouse |
| Conjugate: | Unconjugated |
Anti-mCherry Antibody (A85306) | |
10µl - 100µl/$190 – $5453 Citations | |
| Description: | Rabbit polyclonal antibody to mCherry. |
| Applications: | WB, ICC/IF, IHC |
| Isotype: | IgG |
| Conjugate: | Unconjugated |
Anti-beta III Tubulin Antibody [TU-20] (A86691) | |
10µg - 100µg/$200 – $3901 Citation | |
| Description: | Mouse monoclonal [TU-20] antibody to beta III Tubulin. |
| Applications: | Flow Cytometry (Intracellular), WB, IHC-P, ICC |
| Reactivity: | Canine, Mouse, Rat, Porcine, Human |
| Conjugate: | Unconjugated |
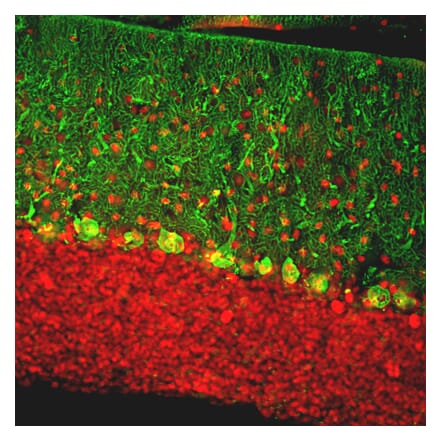
Anti-mCherry Antibody [1C51] (A85305) | |
10µl - 100µl/$190 – $545 | |
| Description: | Mouse monoclonal [1C51] antibody to mCherry. |
| Applications: | WB, ICC/IF, IHC |
| Isotype: | IgG2a |
| Conjugate: | Unconjugated |
Goat Anti-Rabbit IgG H&L Antibody (HRP) (A17345) | |
100µl/$95 | |
| Description: | Goat anti-rabbit IgG H&L (HRP) secondary antibody. |
| Applications: | WB, IHC, ELISA |
| Reactivity: | Rabbit |
| Conjugate: | HRP |
Anti-LAMP2 Antibody [H4B4] - BSA and Azide free (A86605) | |
100µg/$340 | |
| Description: | Mouse monoclonal [H4B4] antibody to LAMP2. |
| Applications: | Flow Cytometry, WB, IHC-P, IHC-Fr, ICC |
| Reactivity: | Human |
| Conjugate: | Unconjugated |
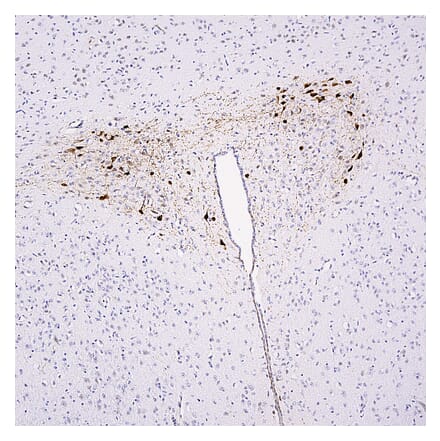
Anti-Ubiquitin Antibody [Ubi-1] (A85456) | |
10µl - 100µl/$190 – $545 | |
| Description: | Mouse monoclonal [Ubi-1] antibody to Ubiquitin. |
| Applications: | WB, ICC/IF, IHC |
| Reactivity: | Human, Monkey, Horse, Bovine, Porcine, Chicken, Rat, Mouse, Danio, Drosophila, C. elegans |
| Conjugate: | Unconjugated |
Anti-HA Tag Antibody [HA.C5] (A85278) | |
10µg - 200µg/$200 – $5653 Citations | |
| Description: | Mouse monoclonal (HA.C5) antibody to HA Tag. |
| Applications: | Dot, ELISA, IP, IS, WB |
| Isotype: | IgG3 |
| Conjugate: | Unconjugated |
Anti-GAPDH Antibody [GA1R] (A85271) | |
10µg - 200µg/$200 – $6202 Citations | |
| Description: | Mouse monoclonal (GA1R) antibody to GAPDH. |
| Applications: | Dot, ELISA, IS, WB |
| Reactivity: | BL-21 Bacteria, Chicken, Hamster, Human, Mouse, Rat, Rabbit, Saccharomyces cerevisiae, Sf9 Insect |
| Conjugate: | Unconjugated |
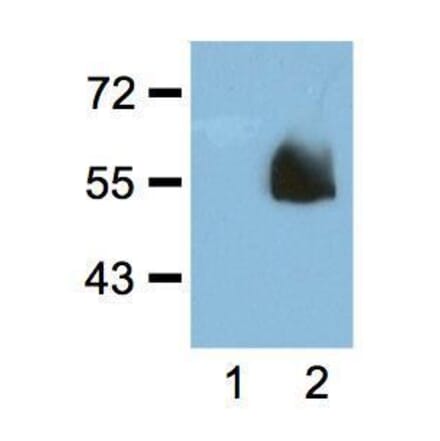
Anti-LAMP1 Antibody [H4A3] - BSA and Azide free (A86537) | |
100µg/$340 | |
| Description: | Mouse monoclonal [H4A3] antibody to LAMP1. |
| Applications: | Flow Cytometry, WB, IHC-P, IHC-Fr, ICC |
| Reactivity: | Human, Non-Human Primates, Mouse |
| Conjugate: | Unconjugated |
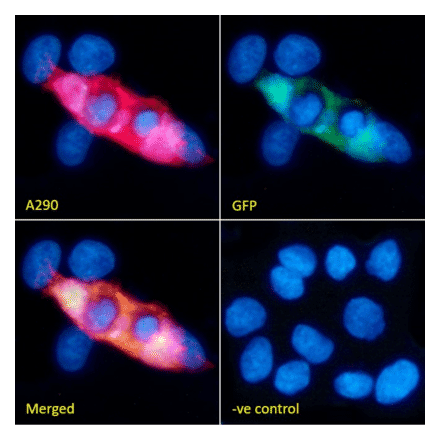
Anti-GFP Antibody (A290) | |
50µl/$455 | |
| Description: | Rabbit polyclonal antibody to GFP. |
| Applications: | WB, ELISA, ICC/IF, Flow Cytometry, IHC-Fr, IHC-P, IP, Electron Microscopy |
| Reactivity: | Species independent |
| Conjugate: | Unconjugated |
Anti-Iba1 Antibody (A104332) | |
10µl - 100µl/$190 – $5451 Citation | |
| Description: | Rabbit polyclonal antibody to Iba1. |
| Applications: | WB, ICC/IF, IHC |
| Reactivity: | Human, Rat, Mouse |
| Conjugate: | Unconjugated |
Anti-Transgelin Antibody (A83697) | |
100µg/$495 | |
| Description: | Goat polyclonal antibody to Transgelin. |
| Applications: | ELISA, WB |
| Reactivity: | Human |
| Conjugate: | Unconjugated |
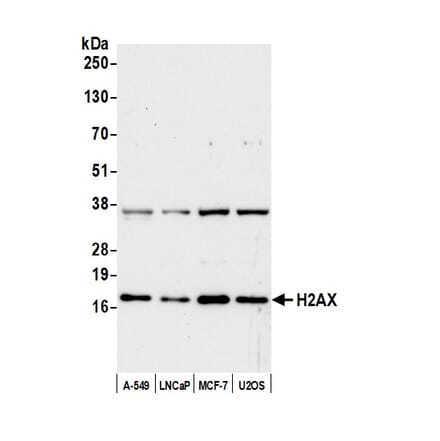
Anti-Histone H2A.X Antibody (A295248) | |
100µl/$545 | |
| Description: | Rabbit polyclonal antibody to Histone H2A.X. |
| Applications: | WB, IHC |
| Reactivity: | Human, Mouse |
| Conjugate: | Unconjugated |
Anti-Vimentin Antibody [VI-10] (A86652) | |
10µg - 100µg/$200 – $3802 Citations | |
| Description: | Mouse monoclonal [VI-10] antibody to Vimentin. |
| Applications: | IP, WB, IHC-P, ICC |
| Reactivity: | Porcine, Chicken, Rat, Human, Mouse |
| Conjugate: | Unconjugated |
Anti-alpha Tubulin Antibody [YOL1/34] (A254436) | |
10µg - 100µg/$200 – $340 | |
| Description: | Rat monoclonal [YOL1/34] antibody to alpha Tubulin. |
| Applications: | WB, IHC-P, ICC, Flow Cytometry, ELISA, IHC-Fr |
| Reactivity: | Mouse, Rat, Mammalian, Avian, Saccharomyces cerevisiae, Human |
| Conjugate: | Unconjugated |
Goat Anti-Mouse IgG H&L Antibody (HRP) (A17352) | |
100µl/$952 Citations | |
| Description: | Goat anti-mouse IgG H&L (HRP) secondary antibody. |
| Applications: | WB, ELISA |
| Reactivity: | Mouse |
| Conjugate: | HRP |
Anti-gamma Histone H2A.X Antibody (A295244) | |
100µl/$545 | |
| Description: | Rabbit polyclonal antibody to gamma Histone H2A.X. |
| Applications: | WB, IHC, ICC/IF |
| Reactivity: | Human, Mouse |
| Conjugate: | Unconjugated |
Anti-Cytokeratin 18 Antibody [C-04] (A86656) | |
10µg - 100µg/$200 – $355 | |
| Description: | Mouse monoclonal [C-04] antibody to Cytokeratin 18. |
| Applications: | IHC-P, IP, WB, ICC, ELISA, Flow Cytometry (Intracellular) |
| Reactivity: | Mammalian |
| Conjugate: | Unconjugated |
Anti-GFAP Antibody (A83720) | |
100µg/$495 | |
| Description: | Goat polyclonal antibody to GFAP. |
| Applications: | ELISA, WB |
| Reactivity: | Human, Mouse, Rat |
| Conjugate: | Unconjugated |
Anti-NeuN Antibody (A85403) | |
10µl - 100µl/$190 – $545 | |
| Description: | Rabbit polyclonal antibody to NeuN. |
| Applications: | WB, ICC/IF, IHC |
| Reactivity: | Human, Rat, Mouse |
| Conjugate: | Unconjugated |
Anti-Laminin Antibody (A85395) | |
10µl - 100µl/$190 – $545 | |
| Description: | Rabbit polyclonal antibody to Laminin. |
| Applications: | WB, ICC/IF, IHC |
| Reactivity: | Human, Rat, Mouse |
| Conjugate: | Unconjugated |
Anti-Myc Tag Antibody (A295120) | |
100µg/$465 | |
| Description: | Goat polyclonal antibody to Myc Tag. |
| Applications: | WB, IP, ICC, ELISA, ChIP |
| Isotype: | IgG |
| Conjugate: | Unconjugated |
Anti-Calbindin Antibody (A85359) | |
10µl - 100µl/$190 – $5451 Citation | |
| Description: | Chicken polyclonal antibody to Calbindin. |
| Applications: | WB, ICC/IF, IHC |
| Reactivity: | Human, Bovine, Rat, Mouse |
| Conjugate: | Unconjugated |
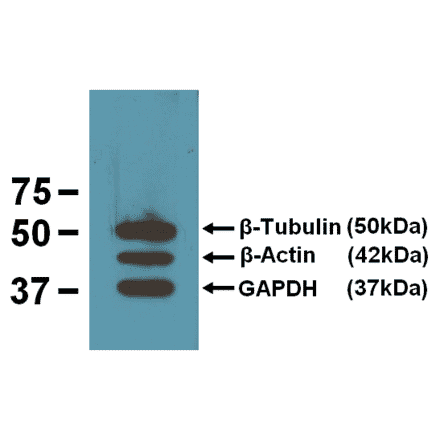
Anti-beta Actin Antibody [BA3R] (A85272) | |
10µg - 200µg/$200 – $5651 Citation | |
| Description: | Mouse monoclonal (BA3R) antibody to beta Actin. |
| Applications: | Dot, ELISA, IS, WB |
| Reactivity: | Chicken, Human, Mouse, Rat, Rabbit |
| Conjugate: | Unconjugated |
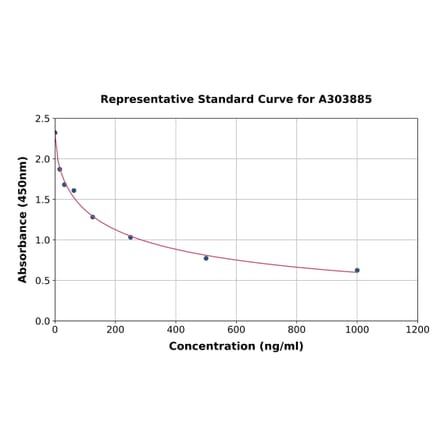
FLAG Tag ELISA Kit (A303885) | |
96T/$770 | |
| Applications: | Competitive ELISA |
| Reactivity: | Universal |
| Sample Type: | Serum, plasma, tissue homogenates, and other biological fluids. |
| Range: | 15.625-1000 ng/ml |
Anti-alpha Smooth Muscle Actin Antibody (A82445) | |
100µg/$4952 Citations | |
| Description: | Goat polyclonal antibody to alpha Smooth Muscle Actin. |
| Applications: | ELISA, WB |
| Reactivity: | Human, Mouse, Rat |
| Conjugate: | Unconjugated |
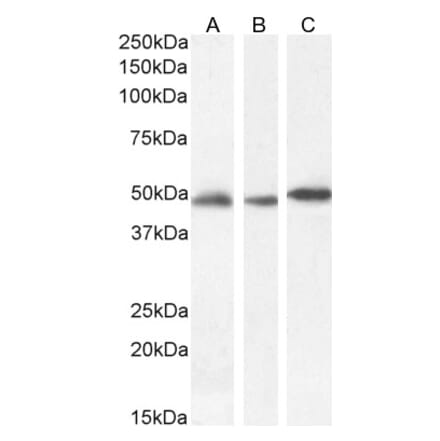
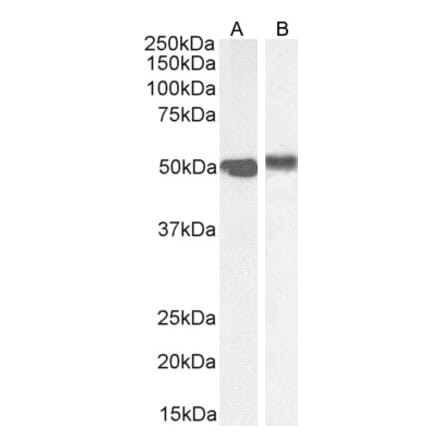
Anti-DNMT1 Antibody [60B1220.1] (A304715) | |
100µg/$520 | |
| Description: | Mouse monoclonal [60B1220.1] antibody to DNMT1. |
| Applications: | WB, IHC, IP, ChIP |
| Reactivity: | Human, Mouse, Fish, Zebrafish |
| Conjugate: | Unconjugated |
Anti-FOXA1 Antibody (A83938) | |
100µg/$495 | |
| Description: | Goat polyclonal antibody to FOXA1. |
| Applications: | ELISA, WB, IF, Flow Cytometry |
| Reactivity: | Human, Mouse |
| Conjugate: | Unconjugated |
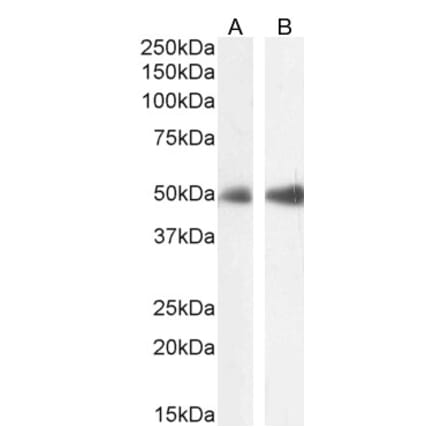
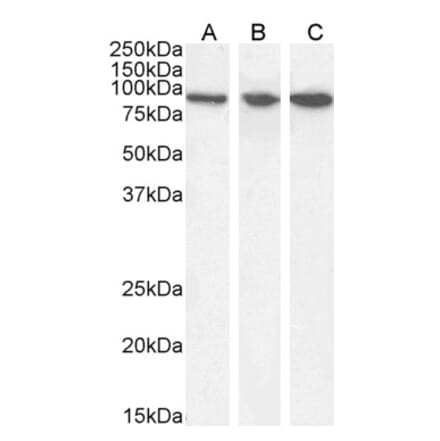
Anti-LAMP2 Antibody [GL2A7] (A304947) | |
100µg/$480 | |
| Description: | Rat monoclonal [GL2A7] antibody to LAMP2. |
| Applications: | WB, ICC/IF, IP |
| Reactivity: | Human, Mouse, Rabbit |
| Conjugate: | Unconjugated |
Anti-VPS35 Antibody (A83699) | |
100µg/$4951 Citation | |
| Description: | Goat polyclonal antibody to VPS35. |
| Applications: | ELISA, WB, IHC, IF |
| Reactivity: | Human, Mouse, Rat |
| Conjugate: | Unconjugated |
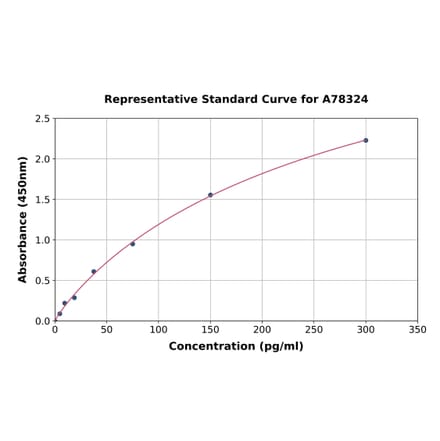
Human IL-6 ELISA Kit (A78324) | |
96T/$570 | |
| Applications: | Sandwich ELISA |
| Reactivity: | Human |
| Sample Type: | Serum, plasma, tissue homogenates, and other biological fluids. |
| Range: | 4.688-300 pg/ml |
Anti-NQO1 Antibody (A82457) | |
100µg/$495 | |
| Description: | Goat polyclonal antibody to NQO1. |
| Applications: | ELISA, WB, IF, IHC |
| Reactivity: | Human, Rat |
| Conjugate: | Unconjugated |
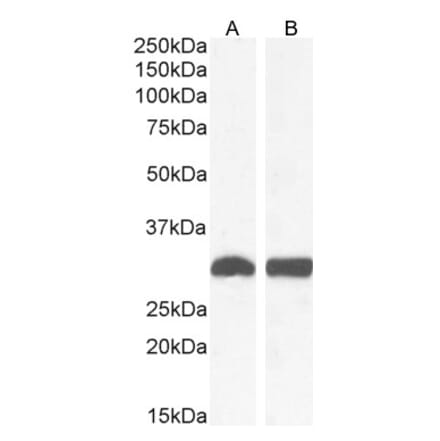
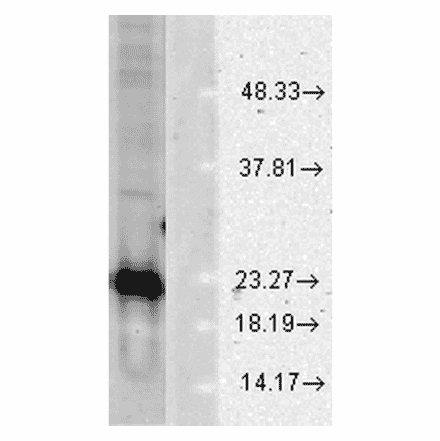
Anti-Superoxide Dismutase 1 Antibody (A305177) | |
100µg/$540 | |
| Description: | Rabbit polyclonal antibody to Superoxide Dismutase 1. |
| Applications: | WB, IHC, IP, ELISA |
| Reactivity: | Human, Rat, Mouse, Bovine, Monkey, Invertebrate, Coral, Canine, Hamster, Porcine, Rabbit, Sheep, Xenopus, Mollusk, Fish |
| Conjugate: | Unconjugated |
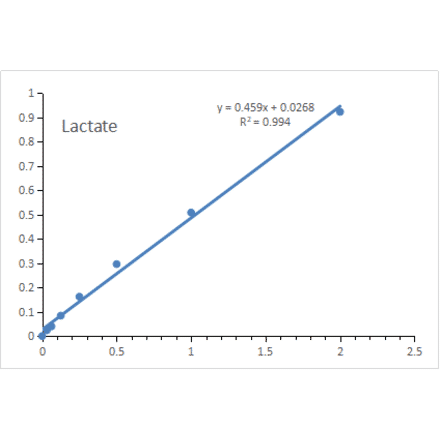
Lactate Assay Kit (A319694) | |
48T - 5 x 96T/$150 – $485 | |
| Description: | This Lactate Assay Kit is a quantitative assay kit for measuring Lactate in animal and plant tissues... |
| Applications: | Functional Studies |
| Range: | 0.0313-2 mM |
| Sensitivity: | 0.0313 mM |
Anti-CD63 Antibody [MEM-259] (A85976) | |
10µg - 100µg/$200 – $355 | |
| Description: | Mouse monoclonal [MEM-259] antibody to CD63. |
| Applications: | Flow Cytometry, IP, IHC-P, ICC |
| Reactivity: | Human |
| Conjugate: | Unconjugated |
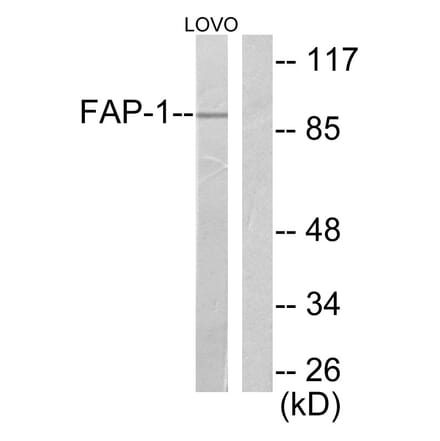
Anti-FAP-1 Antibody (A95883) | |
10µg - 100µg/$190 – $475 | |
| Description: | Rabbit polyclonal antibody to FAP-1. |
| Applications: | WB, IHC, ELISA |
| Reactivity: | Human, Mouse, Rat |
| Conjugate: | Unconjugated |
Anti-NF-H Antibody (A85336) | |
10µl - 100µl/$190 – $545 | |
| Description: | Rabbit polyclonal antibody to NF-H. |
| Applications: | WB, ICC/IF, IHC |
| Reactivity: | Human, Rat, Mouse, Bovine, Porcine, Horse |
| Conjugate: | Unconjugated |
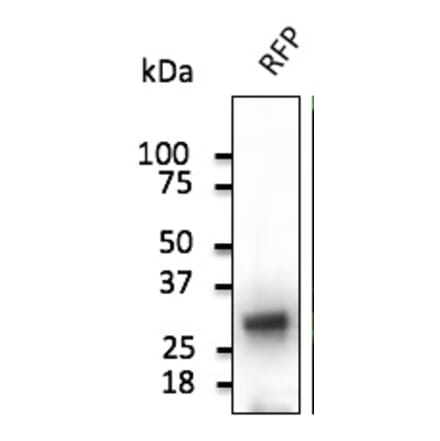
Anti-RFP Antibody (A121675) | |
300µg/$555 | |
| Description: | Goat polyclonal antibody to RFP. |
| Applications: | WB, IF, IHC-P, IHC-Fr, IEM |
| Reactivity: | Red Fluorescent Protein (dsRed), mCherry, mOrange, mPlum, mRFP, mStrawberry, tdTomato |
| Conjugate: | Unconjugated |
Anti-FOXL2 Antibody (A83945) | |
100µg/$495 | |
| Description: | Goat polyclonal antibody to FOXL2. |
| Applications: | ELISA, WB, IHC |
| Reactivity: | Human, Mouse |
| Conjugate: | Unconjugated |
Showing 1-50 of 118,610 products
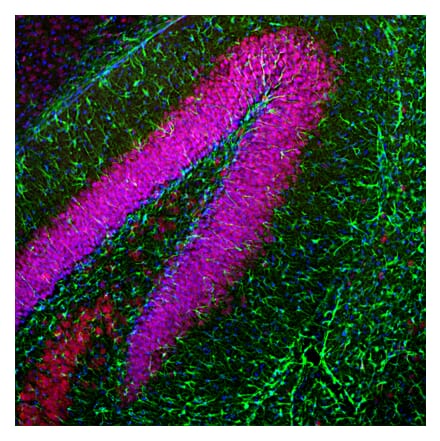
![Flow Cytometry - Anti-PCNA Antibody [PC10] (A86877) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86878_948.jpg?profile=product_search)

![Immunofluorescence - Anti-Fibrillarin Antibody [38F3] (A85370) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85370_1.jpg?profile=product_search)
![Immunofluorescence - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_1.jpg?profile=product_search)
![Immunocytochemistry - Anti-beta III Tubulin Antibody [TU-20] (A86689) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86691_816.jpg?profile=product_search)
![Immunohistochemistry - Anti-mCherry Antibody [1C51] (A85305) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85305_1.jpg?profile=product_search)

![Immunohistochemistry - Anti-Ubiquitin Antibody [Ubi-1] (A85456) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85456_1.jpg?profile=product_search)
![Flow Cytometry - Anti-CD107a Antibody [H4A3] - BSA and Azide free (A86536) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86537_711.jpg?profile=product_search)

![Immunocytochemistry - Anti-Vimentin Antibody [VI-10] (A86650) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86652_789.jpg?profile=product_search)
![Immunocytochemistry - Anti-alpha Tubulin Antibody [YOL1/34] (A254435) - Antibodies.com](https://cdn.antibodies.com/image/catalog/254/A254436_1.jpg?profile=product_search)
![Immunohistochemistry - Anti-Cytokeratin 18 Antibody [C-04] (A86656) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86656_794.jpg?profile=product_search)

![Western Blot - Anti-DNMT1 Antibody [60B1220.1] (A304715) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304715_1.png?profile=product_search)
![Western Blot - Anti-LAMP2 Antibody [GL2A7] (A304947) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304947_1.png?profile=product_search)
![Flow Cytometry - Anti-CD63 Antibody [MEM-259] (A85975) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85976_346.jpg?profile=product_search)