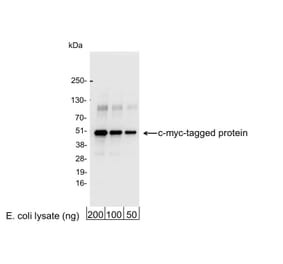
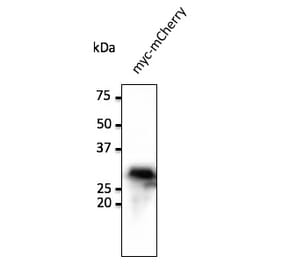
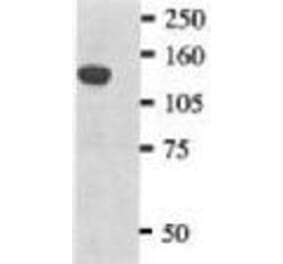
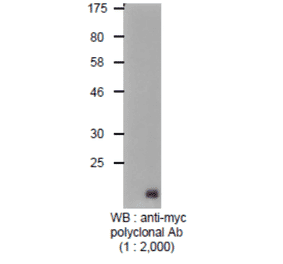
Unconjugated
Repair of interstrand crosslinks by the Fanconi anemia (FA) pathway requires both monoubiquitination and de-ubiquitination of the FANCI/FANCD2 (FANCI/D2) complex. In the standing model, the phosphorylation of six sites in the FANCI S/TQ cluster domain occurs upstream of, and promotes, FANCI/D2 monoubiquitination. We generated phospho-specific antibodies against three different S/TQ cluster sites (serines 556, 559, and 565) on human FANCI and found that, in contrast to the standing model, distinct FANCI sites were phosphorylated either predominantly upstream (ubiquitination independent; serine 556) or downstream (ubiquitination-linked; serines 559 and 565) of FANCI/D2 monoubiquitination. Ubiquitination-linked FANCI phosphorylation inhibited FANCD2 de-ubiquitination and bypassed the need to de-ubiquitinate FANCD2 to achieve effective interstrand crosslink repair. USP1 depletion suppressed ubiquitination-linked FANCI phosphorylation despite increasing FANCI/D2 monoubiquitination, providing an explanation of why FANCD2 de-ubiquitination is important for function of the FA pathway. Our work results in a refined model of how FANCI phosphorylation activates the FANCI/D2 complex.
Mammalian parental genomes contribute differently to early embryonic development. Before activation of the zygotic genome, the maternal genome provides all transcripts and proteins required for the transition from a highly specialized oocyte to a pluripotent embryo. Depletion of these maternally-encoded transcripts frequently results in failure of preimplantation embryonic development, but their functions in this process are incompletely understood. We found that female mice lacking NLRP2 are subfertile because of early embryonic loss and the production of fewer offspring that have a wide array of developmental phenotypes and abnormal DNA methylation at imprinted loci. By demonstrating that NLRP2 is a member of the subcortical maternal complex (SCMC), an essential cytoplasmic complex in oocytes and preimplantation embryos with poorly understood function, we identified imprinted postzygotic DNA methylation maintenance, likely by directing subcellular localization of proteins involved in this process, such as DNMT1, as a new crucial role of the SCMC for mammalian reproduction.
Retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) are critical cytosolic sensors that trigger the production of interferons (IFNs). Though their recognition functions are well identified, their unique roles in the downstream signal transduction remain to be elucidated. Herein, we report the differential effect between grass carp (Ctenopharyngodon idella) MDA5 (CiMDA5) and CiRIG-I on the production of various IFNs upon grass carp reovirus (GCRV) infection in C. idella kidney (CIK) cell line. In CIK cells, grass carp IFN1 (CiIFN1) and CiIFN3 are relatively highly expressed while CiIFN2 and CiIFN4 are relatively slightly expressed. Following GCRV infection, CiMDA5 induces a more extensive type I IFN response than CiRIG-I. Further investigation reveals that both CiMDA5 and CiRIG-I facilitate the expression and total phosphorylation levels of grass carp IFN regulatory factor (IRF) 3 (CiIRF3) and CiIRF7 upon GCRV infection or poly(I:C) stimulation. However, the difference is that CiRIG-I decreases the threonine phosphorylation level of CiIRF7. As a consequence, CiMDA5 enhances the heterodimerization of CiIRF3 and CiIRF7 and homodimerization of CiIRF7, whereas CiRIG-I facilitates the heterodimerization but attenuates homodimerization of CiIRF7. Moreover, the present study suggests that CiIRF3 and CiIRF7 heterodimers and CiIRF7 homodimers are able to induce more extensive IFN-I responses than CiIRF3 homodimers under GCRV infection. Additionally, CiMDA5 induces a stronger type II IFN (IFN-II) response against GCRV infection than CiRIG-I. Collectively, these results demonstrate that CiMDA5 plays a more potent role than CiRIG-I in IFN response to GCRV infection through differentially regulating the phosphorylation and dimerization of CiIRF3 and CiIRF7.
Targeting self-renewal is an important goal in cancer therapy and recent studies have focused on Notch signalling in the maintenance of stemness of glioma stem cells (GSCs). Understanding cancer-specific Notch regulation would improve specificity of targeting this pathway. In this study, we find that Notch1 activation in GSCs specifically induces expression of the lncRNA, TUG1. TUG1 coordinately promotes self-renewal by sponging miR-145 in the cytoplasm and recruiting polycomb to repress differentiation genes by locus-specific methylation of histone H3K27 via YY1-binding activity in the nucleus. Furthermore, intravenous treatment with antisense oligonucleotides targeting TUG1 coupled with a drug delivery system induces GSC differentiation and efficiently represses GSC growth in vivo. Our results highlight the importance of the Notch-lncRNA axis in regulating self-renewal of glioma cells and provide a strong rationale for targeting TUG1 as a specific and potent therapeutic approach to eliminate the GSC population.
The over-expression of regulator of calcineurin 1 isoform 1 (RCAN1.1) has been implicated in mitochondrial dysfunctions of Alzheimer's disease; however, the mechanism linking RCAN1.1 over-expression and the mitochondrial dysfunctions remains unknown. In this study, we use human neuroblastoma SH-SY5Y cells stably expressing RCAN1.1S and rat primary neurons infected with RCAN1.1S expression lentivirus to study the association of RCAN1 with mitochondrial functions. Our study here showed that the over-expression of RCAN1.1S remarkably up-regulates the expression of adenine nucleotide translocator (ANT1) by stabilizing ANT1 mRNA. The increased ANT1 level leads to accelerated ATP-ADP exchange rate, more Ca2+ -induced mitochondrial permeability transition pore opening, increased cytochrome c release, and eventually cell apoptosis. Furthermore, knockdown of ANT1 expression brings these mitochondria perturbations caused by RCAN1.1S back to normal. The effect of RCAN1.1S on ANT1 was independent of its inhibition on calcineurin. This study elucidated a novel function of RCAN1 in mitochondria and provides a molecular basis for the RCAN1.1S over-expression-induced mitochondrial dysfunctions and neuronal apoptosis.
Branched-chain amino acid transaminase 1 (BCAT1) has been associated with numerous types of tumors; however, few previous studies have evaluated the expression and role of BCAT1 in hepatocellular carcinoma (HCC). In the present study, the expression of BCAT1 was detected by reverse transcription-quantitative polymerase chain reaction and immunoblotting in six HCC cell lines and 74 pairs of HCC and adjacent non-cancerous liver tissues. In addition, the correlation between the expression levels of c-Myc and BCAT1 was analyzed using immunohistochemistry. Furthermore, RNA silencing was performed using c-Myc-specific or BCAT1-specific small interfering RNA, after which wound healing and Transwell cell invasion assays were performed. Finally, the clinicopathological characteristics of BCAT1 in patients with HCC were analyzed. It was shown that the expression of BCAT1 was significantly higher in HCC tissues compared with adjacent non-tumor tissues (P<0.001), and in HCC cell lines compared within the L-02 hepatic cell line (P<0.001). In addition, immunohistochemical analyses indicated that the expression of BCAT1 was positively correlated with c-Myc (r=0.706, P<0.001). BCAT1 expression was shown to be downregulated in c-Myc-knockdown cells, and silencing of BCAT1 expression reduced the invasion and migration of HCC cells. Furthermore, a clinical analysis indicated that BCAT1 expression in HCC tissues was significantly associated with the tumor-node-metastasis stage, tumor number and tumor differentiation (all P<0.05), and that BCAT1 was able to predict the 5-year survival and disease-free survival rates of patients with HCC (both P<0.001). The results of the present study suggested that BCAT1 expression is upregulated in patients with HCC, and that BCAT1 may serve as a potential molecular target for the diagnosis and treatment of HCC.
Seeds maintain a dormant state to withstand adverse conditions and germinate when conditions become favourable to give rise to a new generation of flowering plants. Seed dormancy and germination are tightly controlled by internal and external signals. Although phytochrome photoreceptors are proposed to regulate primary seed dormancy, the underlying molecular mechanism remains elusive. Here we show that the REVEILLE1 (RVE1) and RVE2 transcription factors promote primary seed dormancy and repress red/far-red-light-reversible germination downstream of phytochrome B (phyB) in Arabidopsis thaliana. RVE1 and RVE2 expression is downregulated after imbibition and by phyB. RVE1 directly binds to the promoter of GIBBERELLIN 3-OXIDASE 2, inhibits its transcription and thus suppresses the biosynthesis of bioactive gibberellins. In addition, DELAY OF GERMINATION 1 also acts downstream of phyB. This study identifies a signalling pathway that integrates environmental light input with internal factors to control both seed dormancy and germination.
The trimethylation of histone H3 on lysine 9 (H3K9me3) - a mark recognized by HP1 that depends on the Suv39h lysine methyltransferases (KMTs) - has provided a basis for the reader/writer model to explain HP1 accumulation at pericentric heterochromatin in mammals. Here, we identify the Suv39h1 paralog, as a unique enhancer of HP1α sumoylation both in vitro and in vivo. The region responsible for promoting HP1α sumoylation (aa1-167) is distinct from the KMT catalytic domain and mediates binding to Ubc9. Tethering the 1-167 domain of Suv39h1 to pericentric heterochromatin, but not mutants unable to bind Ubc9, accelerates the de novo targeting of HP1α to these domains. Our results establish an unexpected feature of Suv39h1, distinct from the KMT activity, with a major role for heterochromatin formation. We discuss how linking Suv39h1 to the SUMO pathway provides conceptual implications for our general view on nuclear domain organization and physiological functions.
Here, we describe a new strategy that allows the rapid and efficient engineering of mono and multispecific trivalent antibodies. By fusing single-domain antibodies from camelid heavy-chain-only immunoglobulins (VHHs) to the N-terminus of a human collagen XVIII trimerization domain (TIE(XVIII)) we produced monospecific trimerbodies that were efficiently secreted as soluble functional proteins by mammalian cells. The purified VHH-TIE(XVIII) trimerbodies were trimeric in solution and exhibited excellent antigen binding capacity. Furthermore, by connecting with two additional glycine-serine-based linkers three VHH-TIE(XVIII) modules on a single polypeptide chain, we present an approach for the rational design of multispecific tandem trimerbodies with defined stoichiometry and controlled orientation. Using this technology we report here the construction and characterization of a tandem VHH-based trimerbody capable of simultaneously binding to three different antigens: carcinoembryonic antigen (CEA), epidermal growth factor receptor (EGFR) and green fluorescence protein (GFP). Multispecific tandem VHH-based trimerbodies were well expressed in mammalian cells, had good biophysical properties and were capable of simultaneously binding their targeted antigens. Importantly, these antibodies were very effective in inhibiting the proliferation of human epidermoid carcinoma A431 cells. Multispecific VHH-based trimerbodies are therefore ideal candidates for future applications in various therapeutic areas.
Both p53-related p63 and c-Myc are transcription factors playing key roles in cell proliferation, survival, development and tumorigenesis. In the present study, we identified that MM1, a c-Myc inhibitor, specifically binds to C-termini of p63α (including ΔNp63α and TAp63α). Further study demonstrates that p63α facilitates MM1 protein degradation via proteasomal pathway, resulting in elevation of c-Myc transactivation activity. Knockdown of ΔNp63α leads to decrease in c-Myc protein levels, concomitant with reduced expression of CDK4 and Cyclin D1, and impaired cell cycle progression, both of which are effectively reversed by simultaneous knockdown of MM1. Moreover, expression of p63 and CDK4 is concomitantly up-regulated in B-cell acute lymphoblastic leukemia. Together, this study reveals a novel crosstalk between p63 and c-Myc that may play an important role in cell cycle progression and tumorigenesis.
The MORC family of GHKL ATPases are an enigmatic class of proteins with diverse chromatin related functions. In Arabidopsis, AtMORC1, AtMORC2, and AtMORC6 act together in heterodimeric complexes to mediate transcriptional silencing of methylated DNA elements. Here, we studied Arabidopsis AtMORC4 and AtMORC7. We found that, in contrast to AtMORC1,2,6, they act to suppress a wide set of non-methylated protein-coding genes that are enriched for those involved in pathogen response. Furthermore, atmorc4 atmorc7 double mutants show a pathogen response phenotype. We found that AtMORC4 and AtMORC7 form homomeric complexes in vivo and are concentrated in discrete nuclear bodies adjacent to chromocenters. Analysis of an atmorc1,2,4,5,6,7 hextuple mutant demonstrates that transcriptional de-repression is largely uncoupled from changes in DNA methylation in plants devoid of MORC function. However, we also uncover a requirement for MORC in both DNA methylation and silencing at a small but distinct subset of RNA-directed DNA methylation target loci. These regions are characterized by poised transcriptional potential and a low density of sites for symmetric cytosine methylation. These results provide insight into the biological function of MORC proteins in higher eukaryotes.
In the central and peripheral nervous system, the assembly of KCNQ3 with KCNQ2 as mostly heteromers, but also homomers, underlies "M-type" currents, a slowly-activating voltage-gated K+ current that plays a dominant role in neuronal excitability. KCNQ3 homomers yield much smaller currents compared to KCNQ2 or KCNQ4 homomers and KCNQ2/3 heteromers. This smaller current has been suggested to result either from divergent channel surface expression or from a pore that is more unstable in KCNQ3. Channel surface expression has been shown to be governed by the distal part of the C-terminus in which helices C and D are critical for channel trafficking and assembly. A sequence alignment of this region in KCNQ channels shows that KCNQ3 possesses a longer linker between helix C and D compared to the other KCNQ subunits. Here, we investigate the role of the extra residues of this linker on KCNQ channel expression. Deletion of these residues increased KCNQ3 current amplitudes. Total internal reflection fluorescence imaging and plasma membrane protein assays suggest that the increase in current is due to a higher surface expression of the channels. Conversely, introduction of the extra residues into the linker between helices C and D of KCNQ4 reduced current amplitudes by decreasing the number of KCNQ4 channels at the plasma membrane. Confocal imaging suggests a higher fraction of channels, which possess the extra residues of helix C-D linker, were retained within the endoplasmic reticulum. Such retention does not appear to lead to protein accumulation and activation of the unfolded protein response that regulates protein folding and maintains endoplasmic reticulum homeostasis. Taken together, we conclude that extra helix C-D linker residues play a role in KCNQ3 current amplitudes by controlling the exit of the channel from the endoplasmic reticulum.
Nuclear factor-κB (NF-κB) has a vital role in cell survival. Inhibition of NF-κB has been proven to be an efficient therapeutic pathway for various cancers. Activation of NF-κB is mainly through serine residues' phosphorylation of inhibitor of κBα (IκBα) by IKK complex. Phosphorylation at tyrosine 42 is an alternative pathway in regulation of IκBα and NF-κB signaling, though little is known about the underlying mechanism. Here we identified regulator of calcineurin 1 (RCAN1) as a novel endogenous inhibitor of NF-κB signaling pathway. RCAN1 can interact with IκBα and affect the phosphorylation of IκBα at tyrosine 42. Overexpression of RCAN1 by adenovirus reduced cell viability in lymphoma Raji cells and restrained the growth of lymphoma transplants in mice. We further found that N terminus 1-103aa of RCAN1 is sufficient to inhibit NF-κB and reduce cell viability of lymphoma cells. Our study implicated a novel therapeutic approach for lymphoma by RCAN1 through inhibition of NF-κB signaling.
Trafficking of cargo through the endosomal system depends on endosomal fusion events mediated by SNARE proteins, Rab-GTPases, and multisubunit tethering complexes. The CORVET and HOPS tethering complexes, respectively, regulate early and late endosomal tethering and have been characterized in detail in yeast where their sequential membrane targeting and assembly is well understood. Mammalian CORVET and HOPS subunits significantly differ from their yeast homologues, and novel proteins with high homology to CORVET/HOPS subunits have evolved. However, an analysis of the molecular interactions between these subunits in mammals is lacking. Here, we provide a detailed analysis of interactions within the mammalian CORVET and HOPS as well as an additional endosomal-targeting complex (VIPAS39-VPS33B) that does not exist in yeast. We show that core interactions within CORVET and HOPS are largely conserved but that the membrane-targeting module in HOPS has significantly changed to accommodate binding to mammalian-specific RAB7 interacting lysosomal protein (RILP). Arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome-associated mutations in VPS33B selectively disrupt recruitment to late endosomes by RILP or binding to its partner VIPAS39. Within the shared core of CORVET/HOPS, we find that VPS11 acts as a molecular switch that binds either CORVET-specific TGFBRAP1 or HOPS-specific VPS39/RILP thereby allowing selective targeting of these tethering complexes to early or late endosomes to time fusion events in the endo/lysosomal pathway.
Extensive reprogramming of cellular energy metabolism is a hallmark of cancer. Despite its importance, the molecular mechanism controlling this tumour metabolic shift remains not fully understood. Here we show that 14-3-3σ regulates cancer metabolic reprogramming and protects cells from tumorigenic transformation. 14-3-3σ opposes tumour-promoting metabolic programmes by enhancing c-Myc poly-ubiquitination and subsequent degradation. 14-3-3σ demonstrates the suppressive impact on cancer glycolysis, glutaminolysis, mitochondrial biogenesis and other major metabolic processes of tumours. Importantly, 14-3-3σ expression levels predict overall and recurrence-free survival rates, tumour glucose uptake and metabolic gene expression in breast cancer patients. Thus, these results highlight that 14-3-3σ is an important regulator of tumour metabolism, and loss of 14-3-3σ expression is critical for cancer metabolic reprogramming. We anticipate that pharmacologically elevating the function of 14-3-3σ in tumours could be a promising direction for targeted anticancer metabolism therapy development in future.
Osteosarcoma (OS) survival rates have plateaued in part due to a lack of new therapeutic options. Here we demonstrate that bromodomain inhibitors (BETi), JQ1, I-BET151, I-BET762, exert potent anti-tumour activity against primary and established OS cell lines, mediated by inhibition of BRD4. Strikingly, unlike previous observations in long-term established human OS cell lines, the antiproliferative activity of JQ1 in primary OS cells was driven by the induction of apoptosis, not cell cycle arrest. In further contrast, JQ1 activity in OS was mediated independently of MYC downregulation. We identified that JQ1 suppresses the transcription factor FOSL1 by displacement of BRD4 from its locus. Loss of FOSL1 phenocopied the antiproliferative effects of JQ1, identifying FOSL1 suppression as a potential novel therapeutic approach for OS. As a monotherapy JQ1 demonstrated significant anti-tumour activity in vivo in an OS graft model. Further, combinatorial treatment approaches showed that JQ1 increased the sensitivity of OS cells to doxorubicin and induced potent synergistic activity when rationally combined with CDK inhibitors. The greater level of activity achieved with the combination of BETi with CDK inhibitors demonstrates the efficacy of this combination therapy. Taken together, our studies show that BET inhibitors are a promising new therapeutic for OS.
DNA-end resection, the generation of single-stranded DNA at DNA double strand break (DSB) ends, is critical for controlling the many cellular responses to breaks. Here we show that the conserved DNA damage checkpoint sliding clamp (the 9-1-1 complex) plays two opposing roles coordinating DSB resection in budding yeast. We show that the major effect of 9-1-1 is to inhibit resection by promoting the recruitment of Rad9(53BP1) near DSBs. However, 9-1-1 also stimulates resection by Exo1- and Dna2-Sgs1-dependent nuclease/helicase activities, and this can be observed in the absence of Rad9(53BP1). Our new data resolve the controversy in the literature about the effect of the 9-1-1 complex on DSB resection. Interestingly, the inhibitory role of 9-1-1 on resection is not observed near uncapped telomeres because less Rad9(53BP1) is recruited near uncapped telomeres. Thus, 9-1-1 both stimulates and inhibits resection and the effects of 9-1-1 are modulated by different regions of the genome. Our experiments illustrate the central role of the 9-1-1 checkpoint sliding clamp in the DNA damage response network that coordinates the response to broken DNA ends. Our results have implications in all eukaryotic cells.
Stress caused by accumulation of misfolded proteins within the endoplasmic reticulum (ER) elicits a cellular unfolded protein response (UPR) aimed at maintaining protein-folding capacity. PERK, a key upstream component, recognizes ER stress via its luminal sensor/transducer domain, but the molecular events that lead to UPR activation remain unclear. Here, we describe the crystal structures of mammalian PERK luminal domains captured in dimeric state as well as in a novel tetrameric state. Small angle X-ray scattering analysis (SAXS) supports the existence of both crystal structures also in solution. The salient feature of the tetramer interface, a helix swapped between dimers, implies transient association. Moreover, interface mutations that disrupt tetramer formation in vitro reduce phosphorylation of PERK and its target eIF2α in cells. These results suggest that transient conversion from dimeric to tetrameric state may be a key regulatory step in UPR activation.
Factor induced reprogramming of fibroblasts is an orchestrated but inefficient process. At the epigenetic level, it results in drastic chromatin changes to erase the existing somatic "memory" and to establish the pluripotent state. Accordingly, alterations of chromatin regulators including Ezh2 influence iPSC generation. While the role of individual transcription factors in resetting the chromatin landscape during iPSC generation is increasingly evident, their engagement with chromatin modulators remains to be elucidated. In the current study, we demonstrate that histone methyl transferase activity of Ezh2 is required for mesenchymal to epithelial transition (MET) during human iPSC generation. We show that the H3K27me3 activity favors induction of pluripotency by transcriptionally targeting the TGF-β signaling pathway. We also demonstrate that the Ezh2 negatively regulates the expression of pro-EMT miRNA's such as miR-23a locus during MET. Unique association of Ezh2 with c-Myc was required to silence the aforementioned circuitry. Collectively, our findings provide a mechanistic understanding by which Ezh2 restricts the somatic programme during early phase of cellular reprogramming and establish the importance of Ezh2 dependent H3K27me3 activity in transcriptional and miRNA modulation during human iPSC generation.
Primary diffuse large B-cell lymphoma of the central nervous system (CNS DLBCL) is a rare, aggressive subtype of DLBCL, the biology of which is poorly understood. Recent studies have suggested a prognostic role of MYC protein expression in systemic DLBCL, but little is known about the frequency and significance of MYC protein expression in CNS DLBCL. Hence, we investigated MYC protein expression profiles of CNS DLBCL and assessed the relationship between MYC expression and a variety of histopathologic, immunophenotypic, genetic, and clinical features. Fifty-nine CNS DLBCL diagnosed at our institution over the past 13 years were evaluated. The majority of cases (80%) showed centroblastic morphology, and 12 (20%) displayed a perivascular pattern of infiltration. According to the Hans criteria, 41 (69%) cases had a non-germinal center B-cell and 18 (31%) had a germinal center B-cell cell-of-origin (COO) phenotype. Mean MYC protein expression was 50% (median: 50%, range: 10-80%). Forty-three cases (73%) showed MYC overexpression (≥ 40%), and 35 (60%) showed MYC/BCL2 coexpression. MYC overexpression was seen in the single case harboring MYC translocation and in the cases showing increased copies of MYC (27%); however, no significant difference in mean MYC expression was seen between groups harboring or lacking MYC aberrations. In our series, age was associated with a significantly increased risk of death, and the perivascular pattern of infiltration was associated with a significantly increased risk of disease progression. Neither MYC expression (with or without BCL2 coexpression) nor other variables, including COO subtype were predictive of clinical outcome. Our findings indicate that the proportion of CNS DLBCL overexpressing MYC is higher compared to systemic DLBCL, and MYC overexpression appears to be independent of genetic MYC abnormalities. Thus, MYC expression and other immunophenotypic markers used for prognostication of systemic DLBCL might not apply to CNS DLBCL due to differences in disease biology.
Our current understanding of how natural genetic variation affects gene expression beyond well-annotated coding genes is still limited. The use of deep sequencing technologies for the study of expression quantitative trait loci (eQTLs) has the potential to close this gap. Here, we generated the first recombinant strain library for fission yeast and conducted an RNA-seq-based QTL study of the coding, non-coding, and antisense transcriptomes. We show that the frequency of distal effects (trans-eQTLs) greatly exceeds the number of local effects (cis-eQTLs) and that non-coding RNAs are as likely to be affected by eQTLs as protein-coding RNAs. We identified a genetic variation of swc5 that modifies the levels of 871 RNAs, with effects on both sense and antisense transcription, and show that this effect most likely goes through a compromised deposition of the histone variant H2A.Z. The strains, methods, and datasets generated here provide a rich resource for future studies.
Planar cell polarity (PCP) signaling has been shown in different studies to either promote or inhibit the malignancy of breast cancer. Using the 21T cell lines, which were derived from an individual patient and represent distinct stages of progression, we show that the prototypical PCP ligand, WNT5A, is expressed highest in 21MT-1 cells (invasive mammary carcinoma) and lowest in 21PT (atypical ductal hyperplasia) and 21NT (ductal carcinoma in situ) cells. Overexpression of WNT5A decreased spherical colony formation and increased invasion and in vivo extravasation only in 21NT cells; whereas overexpression increased migration of both 21PT and 21NT cells. WNT5A overexpression also increased RHOA expression of both cell lines and subsequent RHOA knockdown blocked WNT5A-induced migration, but only partially blocked WNT5A-induced invasion of 21NT cells. PCP can signal through VANGL1 to modulate AP-1 target genes (e.g. MMP3) and induce invasion. VANGL1 knockdown inhibited WNT5A-induced invasion of 21NT cells, but had no effect on WNT5A-induced migration of either 21PT or 21NT cells. WNT5A-induced MMP3 expression was seen only in 21NT cells, an effect that was VANGL1 dependent, but independent of AP-1. We thus provide evidence that PCP signaling can act in a context dependent manner to promote breast cancer progression.
Synapses, the basic units of communication in the brain, require complex molecular machinery for neurotransmitter release and reception. Whereas numerous components of excitatory postsynaptic sites have been identified, relatively few proteins are known that function at inhibitory postsynaptic sites. One such component is neuroligin-2 (NL2), an inhibitory synapse-specific cell surface protein that functions in cell adhesion and synaptic organization via binding to neurexins. In this study, we used a transgenic tandem affinity purification and mass spectrometry strategy to isolate and characterize NL2-associated complexes. Complexes purified from brains of transgenic His6-FLAG-YFP-NL2 mice showed enrichment in the Gene Ontology terms cell-cell signaling and synaptic transmission relative to complexes purified from wild type mice as a negative control. In addition to expected components including GABA receptor subunits and gephyrin, several novel proteins were isolated in association with NL2. Based on the presence of multiple components involved in trafficking and endocytosis, we showed that NL2 undergoes dynamin-dependent endocytosis in response to soluble ligand and colocalizes with VPS35 retromer in endosomes. Inhibitory synapses in brain also present a particular challenge for imaging. Whereas excitatory synapses on spines can be imaged with a fluorescent cell fill, inhibitory synapses require a molecular tag. We find the His6-FLAG-YFP-NL2 to be a suitable tag, with the unamplified YFP signal localizing appropriately to inhibitory synapses in multiple brain regions including cortex, hippocampus, thalamus, and basal ganglia. Altogether, we characterize NL2-associated complexes, demonstrate regulated trafficking of NL2, and provide tools for further proteomic and imaging studies of inhibitory synapses.
Human herpesvirus 6B (HHV-6B) is a ubiquitous pathogen causing lifelong infections in approximately 95% of humans worldwide. To persist within its host, HHV-6B has developed several immune evasion mechanisms, such as latency, during which minimal proteins are expressed, and the ability to disturb innate and adaptive immune responses. The primary cellular targets of HHV-6B are CD4(+) T cells. Previous studies by Flamand et al. (L. Flamand, J. Gosselin, I. Stefanescu, D. Ablashi, and J. Menezes, Blood 85:1263-1271, 1995) reported on the capacity of HHV-6A as well as UV-irradiated HHV-6A to inhibit interleukin-2 (IL-2) synthesis in CD4(+) lymphocytes, suggesting that viral structural components could be responsible for this effect. In the present study, we identified the HHV-6B U54 tegument protein (U54) as being capable of inhibiting IL-2 expression. U54 binds the calcineurin (CaN) phosphatase enzyme, causing improper dephosphorylation and nuclear translocation of NFAT (nuclear factor of activated T cells) proteins, resulting in suboptimal IL-2 gene transcription. The U54 GISIT motif (amino acids 293 to 297), analogous to the NFAT PXIXIT motif, contributed to the inhibition of NFAT activation. IMPORTANCE Human herpesvirus 6A (HHV-6A) and HHV-6B are associated with an increasing number of pathologies. These viruses have developed strategies to avoid the immune response allowing them to persist in the host. Several studies have illustrated mechanisms by which HHV-6A and HHV-6B are able to disrupt host defenses (reviewed in L. Dagna, J. C. Pritchett, and P. Lusso, Future Virol. 8:273-287, 2013, doi:10.2217/fvl.13.7). Previous work informed us that HHV-6A is able to suppress synthesis of interleukin-2 (IL-2), a key immune growth factor essential for adequate T lymphocyte proliferation and expansion. We obtained evidence that HHV-6B also inhibits IL-2 gene expression and identified the mechanisms by which it does so. Our work led us to the identification of U54, a virion-associated tegument protein, as being responsible for suppression of IL-2. Consequently, we have identified HHV-6B U54 protein as playing a role in immune evasion. These results further contribute to our understanding of HHV-6 interactions with its human host and the efforts deployed to ensure its long-term persistence.
Single-stranded DNA (ssDNA) at DNA ends is an important regulator of the DNA damage response. Resection, the generation of ssDNA, affects DNA damage checkpoint activation, DNA repair pathway choice, ssDNA-associated mutation and replication fork stability. In eukaryotes, extensive DNA resection requires the nuclease Exo1 and nuclease/helicase pair: Dna2 and Sgs1(BLM). How Exo1 and Dna2-Sgs1(BLM) coordinate during resection remains poorly understood. The DNA damage checkpoint clamp (the 9-1-1 complex) has been reported to play an important role in stimulating resection but the exact mechanism remains unclear. Here we show that the human 9-1-1 complex enhances the cleavage of DNA by both DNA2 and EXO1 in vitro, showing that the resection-stimulatory role of the 9-1-1 complex is direct. We also show that in Saccharomyces cerevisiae, the 9-1-1 complex promotes both Dna2-Sgs1 and Exo1-dependent resection in response to uncapped telomeres. Our results suggest that the 9-1-1 complex facilitates resection by recruiting both Dna2-Sgs1 and Exo1 to sites of resection. This activity of the 9-1-1 complex in supporting resection is strongly inhibited by the checkpoint adaptor Rad9(53BP1). Our results provide important mechanistic insights into how DNA resection is regulated by checkpoint proteins and have implications for genome stability in eukaryotes.
Gene therapy to achieve in vivo secretion of recombinant anti-CD3 x anti-tumor bispecific antibodies in cancer patients is being explored as a strategy to counterbalance rapid renal elimination, thereby sustaining levels of bispecific antibodies in the therapeutic range. Here, we performed a comparative analysis between single- and two-chain configurations for anti-CD3 x anti-CEA (carcinoembryonic antigen) bispecific antibodies secreted by genetically-modified human cells. We demonstrate that tandem single-chain variable fragment (scFv) antibodies and two-chain diabodies are expressed as soluble secreted proteins with similar yields. However, we found significant differences in their biological functionality (i.e., antigen binding) and in their ability to induce non-specific T cell activation. Whereas single-chain tandem scFvs induced human T cell activation and proliferation in an antigen-independent manner, secreted two-chain diabodies exerted almost no proliferative stimulus when human T cells were cultured alone or in co-cultures with CEA negative cells. Thus, our data suggest that two-chain diabodies are preferable to single-chain tandem scFvs for immunotherapeutic strategies comprising in vivo secretion of bispecific antibodies aiming to recruit and activate anticancer specific lymphocytic effector T cells.
'Gain' of supernumerary copies of the 8q24.21 chromosomal region has been shown to be common in many human cancers and is associated with poor prognosis. The well-characterized myelocytomatosis (MYC) oncogene resides in the 8q24.21 region and is consistently co-gained with an adjacent 'gene desert' of approximately 2 megabases that contains the long non-coding RNA gene PVT1, the CCDC26 gene candidate and the GSDMC gene. Whether low copy-number gain of one or more of these genes drives neoplasia is not known. Here we use chromosome engineering in mice to show that a single extra copy of either the Myc gene or the region encompassing Pvt1, Ccdc26 and Gsdmc fails to advance cancer measurably, whereas a single supernumerary segment encompassing all four genes successfully promotes cancer. Gain of PVT1 long non-coding RNA expression was required for high MYC protein levels in 8q24-amplified human cancer cells. PVT1 RNA and MYC protein expression correlated in primary human tumours, and copy number of PVT1 was co-increased in more than 98% of MYC-copy-increase cancers. Ablation of PVT1 from MYC-driven colon cancer line HCT116 diminished its tumorigenic potency. As MYC protein has been refractory to small-molecule inhibition, the dependence of high MYC protein levels on PVT1 long non-coding RNA provides a much needed therapeutic target.
Cells allocate substantial resources toward monitoring levels of nutrients that can be used for ATP generation by mitochondria. Among the many specialized cell types, neurons are particularly dependent on mitochondria due to their complex morphology and regional energy needs. Here, we report a molecular mechanism by which nutrient availability in the form of extracellular glucose and the enzyme O-GlcNAc Transferase (OGT), whose activity depends on glucose availability, regulates mitochondrial motility in neurons. Activation of OGT diminishes mitochondrial motility. We establish the mitochondrial motor-adaptor protein Milton as a required substrate for OGT to arrest mitochondrial motility by mapping and mutating the key O-GlcNAcylated serine residues. We find that the GlcNAcylation state of Milton is altered by extracellular glucose and that OGT alters mitochondrial motility in vivo. Our findings suggest that, by dynamically regulating Milton GlcNAcylation, OGT tailors mitochondrial dynamics in neurons based on nutrient availability.
Plant cell elongation is controlled by endogenous hormones, including brassinosteroid (BR) and gibberellin (GA), and by environmental factors, such as light/darkness. The molecular mechanisms underlying the convergence of these signals that govern cell growth remain largely unknown. We previously showed that the chromatin-remodeling factor PICKLE/ENHANCED PHOTOMORPHOGENIC1 (PKL/EPP1) represses photomorphogenesis in Arabidopsis thaliana. Here, we demonstrated that PKL physically interacted with PHYTOCHROME-INTERACTING FACTOR3 (PIF3) and BRASSINAZOLE-RESISTANT1 (BZR1), key components of the light and BR signaling pathways, respectively. Also, this interaction promoted the association of PKL with cell elongation-related genes. We found that PKL, PIF3, and BZR1 coregulate skotomorphogenesis by repressing the trimethylation of histone H3 Lys-27 (H3K27me3) on target promoters. Moreover, DELLA proteins interacted with PKL and attenuated its binding ability. Strikingly, brassinolide and GA3 inhibited H3K27me3 modification of histones associated with cell elongation-related loci in a BZR1- and DELLA-mediated manner, respectively. Our findings reveal that the PKL chromatin-remodeling factor acts as a critical node that integrates light/darkness, BR, and GA signals to epigenetically regulate plant growth and development. This work also provides a molecular framework by which hormone signals regulate histone modification in concert with light/dark environmental cues.
The Forkhead transcription factor FOXA2 plays a fundamental role in controlling metabolic homeostasis in the liver during fasting. The precise molecular regulation of FOXA2 in response to nutrients is not fully understood. Here, we studied whether FOXA2 could be controlled at a post-translational level by acetylation. By means of LC-MS/MS analyses, we identified five acetylated residues in FOXA2. Sirtuin family member SIRT1 was found to interact with and deacetylate FOXA2, the latter process being dependent on the NAD+-binding catalytic site of SIRT1. Deacetylation by SIRT1 reduced protein stability of FOXA2 by targeting it towards proteasomal degradation, and inhibited transcription from the FOXA2-driven G6pase and CPT1a promoters. While mutation of the five identified acetylated residues weakly affected protein acetylation and stability, mutation of at least seven additional lysine residues was required to abolish acetylation and reduce protein levels of FOXA2. The importance of acetylation of FOXA2 became apparent upon changes in nutrient levels. The interaction of FOXA2 and SIRT1 was strongly reduced upon nutrient withdrawal in cell culture, while enhanced Foxa2 acetylation levels were observed in murine liver in vivo after starvation for 36 hours. Collectively, this study demonstrates that SIRT1 controls the acetylation level of FOXA2 in a nutrient-dependent manner and in times of nutrient shortage the interaction between SIRT1 and FOXA2 is reduced. As a result, FOXA2 is protected from degradation by enhanced acetylation, hence enabling the FOXA2 transcriptional program to be executed to maintain metabolic homeostasis.
A large and diverse set of proteins, including CST complex, nonsense mediated decay (NMD), and DNA damage response (DDR) proteins, play important roles at the telomere in mammals and yeast. Here, we report that NMD, like the DDR, affects single-stranded DNA (ssDNA) production at uncapped telomeres. Remarkably, we find that the requirement for Cdc13, one of the components of CST, can be efficiently bypassed when aspects of DDR and NMD pathways are inactivated. However, identical genetic interventions do not bypass the need for Stn1 and Ten1, the partners of Cdc13. We show that disabling NMD alters the stoichiometry of CST components at telomeres and permits Stn1 to bind telomeres in the absence of Cdc13. Our data support a model that Stn1 and Ten1 can function in a Cdc13-independent manner and have implications for the function of CST components across eukaryotes.
The homeodomain transcription factor HHEX (hematopoietically expressed homeobox) has been repeatedly linked to type 2 diabetes mellitus (T2DM) using genome-wide association studies. We report here that within the adult endocrine pancreas, Hhex is selectively expressed in the somatostatin-secreting δ cell. Using two mouse models with Hhex deficiency in the endocrine pancreas, we show that Hhex is required for δ-cell differentiation. Decreased somatostatin levels in Hhex-deficient islets cause disrupted paracrine inhibition of insulin release from β cells. These findings identify Hhex as the first transcriptional regulator specifically required for islet δ cells and suggest compromised paracrine control as a contributor to T2DM.
Reactive oxygen species (ROS)-inducing anticancer agents such as phenethylisothiocyanate (PEITC) activate stress pathways for killing cancer cells. Here we demonstrate that PEITC-induced ROS decreased expression of microRNA 27a (miR-27a)/miR-20a:miR-17-5p and induced miR-regulated ZBTB10/ZBTB4 and ZBTB34 transcriptional repressors, which, in turn, downregulate specificity protein (Sp) transcription factors (TFs) Sp1, Sp3, and Sp4 in pancreatic cancer cells. Decreased expression of miR-27a/miR-20a:miR-17-5p by PEITC-induced ROS is a key step in triggering the miR-ZBTB Sp cascade leading to downregulation of Sp TFs, and this is due to ROS-dependent epigenetic effects associated with genome-wide shifts in repressor complexes, resulting in decreased expression of Myc and the Myc-regulated miRs. Knockdown of Sp1 alone by RNA interference also induced apoptosis and decreased pancreatic cancer cell growth and invasion, indicating that downregulation of Sp transcription factors is an important common mechanism of action for PEITC and other ROS-inducing anticancer agents.
Parkinson's disease is a neurodegenerative disorder characterized by Lewy bodies, a pathological hallmark comprised mostly of aggregated alpha synuclein. Accumulating evidence demonstrates the association of smaller oligomeric aggregates to disease etiology and many therapeutic approaches are aimed at inhibiting and reducing the aggregation process. Molecular chaperones and co-chaperones play a key role in protein homeostasis and have potential as therapeutics to inhibit alpha synuclein associated toxicity. Here we use a gene therapy approach to evaluate the applicability of the Hsp70 co-chaperone CHIP (C-terminal Hsp70 interacting protein) as a therapeutic candidate and examine its direct effect on alpha synuclein aggregates in vivo. Utilizing a novel viral vector mediated rat model to directly detect alpha synuclein aggregates, we show that CHIP can mediate the degradation of alpha synuclein aggregates in vivo. However, our studies also reveal that CHIP may potentially degrade tyrosine hydroxylase which would compromise the applicability of CHIP as a therapeutic approach for Parkinson's disease.
The discovery of multipotent neural crest-derived stem cells, named epidermal neural crest stem cells (EPI-NCSC), that persist postnatally in an easy-to-access location-the bulge of hair follicles-opens a spectrum of novel opportunities for patient-specific therapies. We present a detailed characterization of canine EPI-NCSC (cEPI-NCSC) from multiple dog breeds and protocols for their isolation and ex vivo expansion. Furthermore, we provide novel tools for research in canines, which currently are still scarce. In analogy to human and mouse EPI-NCSC, the neural crest origin of cEPI-NCSC is shown by their expression of the neural crest stem cell molecular signature and other neural crest-characteristic genes. Similar to human EPI-NCSC, cEPI-NCSC also expressed pluripotency genes. We demonstrated that cEPI-NCSC can generate all major neural crest derivatives. In vitro clonal analyses established multipotency and self-renewal ability of cEPI-NCSC, establishing cEPI-NCSC as multipotent somatic stem cells. A critical analysis of the literature on canine spinal cord injury (SCI) showed the need for novel treatments and suggested that cEPI-NCSC represent viable candidates for cell-based therapies in dog SCI, particularly for chondrodystrophic dogs. This notion is supported by the close ontological relationship between neural crest stem cells and spinal cord stem cells. Thus, cEPI-NCSC promise to offer not only a potential treatment for canines but also an attractive and realistic large animal model for human SCI. Taken together, we provide the groundwork for the development of a novel cell-based therapy for a condition with extremely poor prognosis and no available effective treatment.
There is emerging evidence that the misfolding of superoxide dismutase 1 (SOD1) may represent a common pathogenic event in both familial and sporadic amyotrophic lateral sclerosis (ALS). To reduce the burden of misfolded SOD1 species in the nervous system, we have tested a novel therapeutic approach based on adeno-associated virus (AAV)-mediated tonic expression of a DNA construct encoding a secretable single-chain fragment variable (scFv) antibody composed of the variable heavy and light chain regions of a monoclonal antibody (D3H5) binding specifically to misfolded SOD1. A single intrathecal injection of the AAV encoding the single-chain antibody in SOD1(G93A) mice at 45 days of age resulted in sustained expression of single-chain antibodies in the spinal cord, and it delayed disease onset and extension of life span by up to 28%, in direct correlation with scFv titers in the spinal cord. The treatment caused attenuation of neuronal stress signals and reduction in levels of misfolded SOD1 in the spinal cord of SOD1(G93A) mice. From these results, we propose that an immunotherapy based on intrathecal inoculation of AAV encoding a secretable scFv against misfolded SOD1 should be considered as potential treatment for ALS, especially for individuals carrying SOD1 mutations.
Control of lipid droplet (LD) nucleation and copy number are critical, yet poorly understood, processes. We use model peptides that shift from the endoplasmic reticulum (ER) to LDs in response to fatty acids to characterize the initial steps of LD formation occurring in lipid-starved cells. Initially, arriving lipids are rapidly packed in LDs that are resistant to starvation (pre-LDs). Pre-LDs are restricted ER microdomains with a stable core of neutral lipids. Subsequently, a first round of "emerging" LDs is nucleated, providing additional lipid storage capacity. Finally, in proportion to lipid concentration, new rounds of LDs progressively assemble. Confocal microscopy and electron tomography suggest that emerging LDs are nucleated in a limited number of ER microdomains after a synchronized stepwise process of protein gathering, lipid packaging, and recognition by Plin3 and Plin2. A comparative analysis demonstrates that the acyl-CoA synthetase 3 is recruited early to the assembly sites, where it is required for efficient LD nucleation and lipid storage.
Cystin is a novel cilia-associated protein that is disrupted in the cpk mouse, a well-characterized mouse model of autosomal recessive polycystic kidney disease (ARPKD). Interestingly, overexpression of the Myc gene is evident in animal models of ARPKD and is thought to contribute to the renal cystic phenotype. Using a yeast two-hybrid approach, the growth suppressor protein necdin, known to modulate Myc expression, was found as an interacting partner of cystin. Deletion mapping demonstrated that the C-terminus of cystin and both termini of necdin are required for their mutual interaction. Speculating that these two proteins may function to regulate gene expression, we developed a luciferase reporter assay and observed that necdin strongly activated the Myc P1 promoter, and cystin did so more modestly. Interestingly, the necdin effect was significantly abrogated when cystin was co-transfected. Chromatin immunoprecipitation and electrophoretic mobility shift assays revealed a physical interaction with both necdin and cystin and the Myc P1 promoter, as well as between these proteins. The data suggest that these proteins likely function in a regulatory complex. Thus, we speculate that Myc overexpression in the cpk kidney results from the dysregulation of the cystin-necdin regulatory complex and c-Myc, in turn, contributes to cystogenesis in the cpk mouse.
Congenital myasthenic syndromes (CMS) are heterogeneous disorders in which the safety margin of neuromuscular transmission is compromised by one or more specific mechanisms. Using Sanger and exome sequencing in a CMS patient, we identified two heteroallelic mutations, p.Glu1233Lys and p.Arg1277His, in LRP4 coding for the postsynaptic low-density lipoprotein receptor-related protein 4. LRP4, expressed on the surface of the postsynaptic membrane of the neuromuscular junction, is a receptor for neurally secreted agrin, and LRP4 bound by agrin activates MuSK. Activated MuSK in concert with Dok-7 stimulates rapsyn to concentrate and anchor AChR on the postsynaptic membrane and interacts with other proteins implicated in the assembly and maintenance of the neuromuscular junction. LRP4 also functions as an inhibitor of Wnt/beta-catenin signaling. The identified mutations in LRP4 are located at the edge of its 3rd beta-propeller domain and decrease binding affinity of LRP4 for both MuSK and agrin. Mutations in the LRP4 3rd beta-propeller domain were previously reported to impair Wnt signaling and cause bone diseases including Cenani-Lenz syndactyly syndrome and sclerosteosis-2. By analyzing naturally occurring and artificially introduced mutations in the LRP4 3rd beta-propeller domain, we show that the edge of the domain regulates the MuSK signaling whereas its central cavity governs Wnt signaling. We conclude that LRP4 is a new CMS disease gene and that the 3rd beta propeller domain of LRP4 mediates the two signaling pathways in a position-specific manner.
The evolutionarily conserved Sgs1/Top3/Rmi1 (STR) complex plays vital roles in DNA replication and repair. One crucial activity of the complex is dissolution of toxic X-shaped recombination intermediates that accumulate during replication of damaged DNA. However, despite several years of study the nature of these X-shaped molecules remains debated. Here we use genetic approaches and two-dimensional gel electrophoresis of genomic DNA to show that Top3, unassisted by Sgs1 and Rmi1, has modest capacities to provide resistance to MMS and to resolve recombination-dependent X-shaped molecules. The X-shaped molecules have structural properties consistent with hemicatenane-related template switch recombination intermediates (Rec-Xs) but not Holliday junction (HJ) intermediates. Consistent with these findings, we demonstrate that purified Top3 can resolve a synthetic Rec-X but not a synthetic double HJ in vitro. We also find that unassisted Top3 does not affect crossing over during double strand break repair, which is known to involve double HJ intermediates, confirming that unassisted Top3 activities are restricted to substrates that are distinct from HJs. These data help illuminate the nature of the X-shaped molecules that accumulate during replication of damaged DNA templates, and also clarify the roles played by Top3 and the STR complex as a whole during the resolution of replication-associated recombination intermediates.
The synaptonemal complex (SC) is a widely conserved structure that mediates the intimate alignment of homologous chromosomes during meiotic prophase and is required for proper homolog segregation at meiosis I. However, fundamental details of SC architecture and assembly remain poorly understood. The coiled-coil protein, Zip1, is the only component whose arrangement within the mature SC of budding yeast has been extensively characterized. It has been proposed that the Small Ubiquitin-like MOdifier, SUMO, plays a role in SC assembly by linking chromosome axes with Zip1's C termini. The role of SUMO in SC structure has not been directly tested, however, because cells lacking SUMO are inviable. Here, we provide direct evidence for SUMO's function in SC assembly. A meiotic smt3 reduction-of-function strain displays reduced sporulation, abnormal levels of crossover recombination, and diminished SC assembly. SC structures are nearly absent when induced at later meiotic time points in the smt3 reduction-of-function background. Using Structured Illumination Microscopy we furthermore determine the position of SUMO within budding yeast SC structure. In contrast to previous models that positioned SUMO near Zip1's C termini, we demonstrate that SUMO lies at the midline of SC central region proximal to Zip1's N termini, within a subdomain called the "central element". The recently identified SUMOylated SC component, Ecm11, also localizes to the SC central element. Finally, we show that SUMO, Ecm11, and even unSUMOylatable Ecm11 exhibit Zip1-like ongoing incorporation into previously established SCs during meiotic prophase and that the relative abundance of SUMO and Ecm11 correlates with Zip1's abundance within SCs of varying Zip1 content. We discuss a model in which central element proteins are core building blocks that stabilize the architecture of SC near Zip1's N termini, and where SUMOylation may occur subsequent to the incorporation of components like Ecm11 into an SC precursor structure.
Pruning that selectively eliminates unnecessary axons/dendrites is crucial for sculpting the nervous system during development. During Drosophila metamorphosis, dendrite arborization neurons, ddaCs, selectively prune their larval dendrites in response to the steroid hormone ecdysone, whereas mushroom body γ neurons specifically eliminate their axon branches within dorsal and medial lobes. However, it is unknown which E3 ligase directs these two modes of pruning. Here, we identified a conserved SCF E3 ubiquitin ligase that plays a critical role in pruning of both ddaC dendrites and mushroom body γ axons. The SCF E3 ligase consists of four core components Cullin1/Roc1a/SkpA/Slimb and promotes ddaC dendrite pruning downstream of EcR-B1 and Sox14, but independently of Mical. Moreover, we demonstrate that the Cullin1-based E3 ligase facilitates ddaC dendrite pruning primarily through inactivation of the InR/PI3K/TOR pathway. We show that the F-box protein Slimb forms a complex with Akt, an activator of the InR/PI3K/TOR pathway, and promotes Akt ubiquitination. Activation of the InR/PI3K/TOR pathway is sufficient to inhibit ddaC dendrite pruning. Thus, our findings provide a novel link between the E3 ligase and the InR/PI3K/TOR pathway during dendrite pruning.
High risk subtype HPV16 early oncoprotein E6 contributes host cell immortalization and transformation through interacting with a number of cellular factors. ING4 is one member of the inhibitor of growth (ING) family of type II tumor suppressors and it has been shown to be involved in regulating p53 function. However, the effect and mechanism of HPV16 E6 on ING4 function remain elusive. In this study, we report HPV16 E6 combines with ING4 in vivo and in vitro. The ING4 induced p53 acetylation and its combining with p53 were attenuated by HPV16 E6 independent of p53 degradation. The enhancing function of ING4 on p53 mediated apoptosis was diminished when HPV16 E6 existed. These findings reveal that ING4 may be a common target of oncogenic viruses for driving host cell carcinogenesis.
Our previous studies on a β1,6-N-acetylglucosaminyltransferase, GnT-IX (GnT-Vb), a homolog of GnT-V, indicated that the enzyme has a broad GlcNAc transfer activity toward N-linked and O-mannosyl glycan core structures and that its brain-specific gene expression is regulated by epigenetic histone modifications. In this study, we demonstrate the existence of an endogenous inhibitory factor for GnT-IX that functions as a key regulator for GnT-IX enzymatic activity in Neuro2a (N2a) cells. We purified this factor from N2a cells and found that it is identical to ectonucleotide pyrophosphatase/phosphodiesterase 3 (ENPP3), as evidenced by mass spectrometry and by the knockdown and overexpression of ENPP3 in cultured cells. Kinetic analyses revealed that the mechanism responsible for the inhibition of GnT-IX caused by ENPP3 is the ENPP3-mediated hydrolysis of the nucleotide sugar donor substrate, UDP-GlcNAc, with the resulting generation of UMP, a potent and competitive inhibitor of GnT-IX. Indeed, ENPP3 knockdown cells had significantly increased levels of intracellular nucleotide sugars and displayed changes in the total cellular glycosylation profile. In addition to chaperones or other known regulators of glycosyltransferases, the ENPP3-mediated hydrolysis of nucleotide sugars would have widespread and significant impacts on glycosyltransferase activities and would be responsible for altering the total cellular glycosylation profile and modulating cellular functions.
In Schizosaccharomyces pombe, heterochromatin spread, which is marked by histone 3 lysine 9 methylation (H3K9me), requires the chromodomains (CDs) of the H3K9 methylase Suv39/Clr4 and the HP1/Swi6 protein. It is unclear how the actions of these two H3K9me-recognizing CDs are coordinated. We find that the intrinsic preference of Suv39/Clr4 is to generate dimethylated H3K9 product. The recognition of pre-existing H3K9me marks by the CD of Suv39/Clr4 stimulates overall catalysis, enabling the accumulation of small amounts of trimethylated product in vivo. Coincidentally, the Suv39/Clr4 CD, unlike the HP1/Swi6 CD, has been shown to prefer the trimethyl state over the dimethyl state. We show that this preference enables efficient heterochromatin spread in vivo by reducing competition with HP1 proteins for the more prevalent dimethyl state. Our results reveal a strategy by which "writers" and "readers" of a chromatin mark exploit different methylation states on the same residue in order to facilitate collaboration and avoid competition.
Previous studies suggested Ataxia-telangiectasia group D complementing gene (ATDC) as an oncogene in many types of cancer. However, its expression and biological functions in non-small cell lung cancer (NSCLC) remain unclear. Herein, we investigated its expression pattern in 109 cases of human NSCLC samples by immunohistochemistry and found that ATDC was overexpressed in 62 of 109 NSCLC samples (56.88%). ATDC overexpression correlated with histological type (p<0.0001), tumor status (p = 0.0227) and histological differentiation (p = 0.0002). Next, we overexpressed ATDC in normal human bronchial epithelial cell line HBE and depleted its expression in NSCLC cell lines A549 and H1299. MTT and colony formation assay showed that ATDC overexpression promoted cell proliferation while its depletion inhibited cell growth. Furthermore, cell cycle analysis showed that ATDC overexpression decreased the percentage of cells in G1 phase and increased the percentage of cells in S phase, while ATDC siRNA treatment increased the G1 phase percentage and decreased the S phase percentage. Further study revealed that ATDC overexpression could up-regulate cyclin D1 and c-Myc expression in HBE cells while its depletion down-regulated cyclin D1 and c-Myc expression in A549 and H1299 cells. In addition, ATDC overexpression was also associated with an increased proliferation index, cyclin D1 and c-Myc expression in human NSCLC samples. Further experiments demonstrated that ATDC up-regulated cyclin D1 and c-Myc expression independent of wnt/β-catenin or p53 signaling pathway. Interestingly, ATDC overexpression increased NF-κB reporter luciferase activity and p-IκB protein level. Correspondingly, NF-κB inhibitor blocked the effect of ATDC on up-regulation of cyclin D1 and c-Myc. In conclusion, we demonstrated that ATDC could promote lung cancer proliferation through NF-κB induced up-regulation of cyclin D1 and c-Myc.
Hypoxia-inducible factor (HIF) 1 and HIF-2 are heterodimeric proteins composed of an oxygen-regulated HIF-1α or HIF-2α subunit, respectively, and a constitutively expressed HIF-1β subunit, which mediate adaptive transcriptional responses to hypoxia. Here, we report that Sirt7 (sirtuin-7) negatively regulates HIF-1α and HIF-2α protein levels by a mechanism that is independent of prolyl hydroxylation and that does not involve proteasomal or lysosomal degradation. The effect of Sirt7 was maintained in the presence of the sirtuin inhibitor nicotinamide and upon deletion or mutation of its deacetylase domain, indicating a non-catalytic function. Knockdown of Sirt7 led to an increase in HIF-1α and HIF-2α protein levels and an increase in HIF-1 and HIF-2 transcriptional activity. Thus, we identify a novel molecular function of Sirt7 as a negative regulator of HIF signaling.
CtIP plays an important role in homologous recombination (HR)-mediated DNA double-stranded break (DSB) repair and interacts with Nbs1 and BRCA1, which are linked to Nijmegen breakage syndrome (NBS) and familial breast cancer, respectively. We identified new CDK phosphorylation sites on CtIP and found that phosphorylation of these newly identified CDK sites induces association of CtIP with the N-terminus FHA and BRCT domains of Nbs1. We further showed that these CDK-dependent phosphorylation events are a prerequisite for ATM to phosphorylate CtIP upon DNA damage, which is important for end resection to activate HR by promoting recruitment of BLM and Exo1 to DSBs. Most notably, this CDK-dependent CtIP and Nbs1 interaction facilitates ATM to phosphorylate CtIP in a substrate-specific manner. These studies reveal one important mechanism to regulate cell-cycle-dependent activation of HR upon DNA damage by coupling CDK- and ATM-mediated phosphorylation of CtIP through modulating the interaction of CtIP with Nbs1, which significantly helps to understand how DSB repair is regulated in mammalian cells to maintain genome stability.
Pannexin1 (Panx1) channels release cytosolic ATP in response to signaling pathways. Panx1 is highly expressed in the central nervous system. We used four antibodies with different Panx1 anti-peptide epitopes to analyze four regions of rat brain. These antibodies labeled the same bands in Western blots and had highly similar patterns of immunofluorescence in tissue culture cells expressing Panx1, but Western blots of brain lysates from Panx1 knockout and control mice showed different banding patterns. Localizations of Panx1 in brain slices were generated using automated wide field mosaic confocal microscopy for imaging large regions of interest while retaining maximum resolution for examining cell populations and compartments. We compared Panx1 expression over the cerebellum, hippocampus with adjacent cortex, thalamus, and olfactory bulb. While Panx1 localizes to the same neuronal cell types, subcellular localizations differ. Two antibodies with epitopes against the intracellular loop and one against the carboxy terminus preferentially labeled cell bodies, while an antibody raised against an N-terminal peptide highlighted neuronal processes more than cell bodies. These labeling patterns may be a reflection of different cellular and subcellular localizations of full-length and/or modified Panx1 channels where each antibody is highlighting unique or differentially accessible Panx1 populations. However, we cannot rule out that one or more of these antibodies have specificity issues. All data associated with experiments from these four antibodies are presented in a manner that allows them to be compared and our claims thoroughly evaluated, rather than eliminating results that were questionable. Each antibody is given a unique identifier through the NIF Antibody Registry that can be used to track usage of individual antibodies across papers and all image and metadata are made available in the public repository, the Cell Centered Database, for on-line viewing, and download.
Despite their classical role as transcriptional repressors, several histone deacetylases, including the baker's yeast Set3/Hos2 complex (Set3C), facilitate gene expression. In the dimorphic human pathogen Candida albicans, the homologue of the Set3C inhibits the yeast-to-filament transition, but the precise molecular details of this function have remained elusive. Here, we use a combination of ChIP-Seq and RNA-Seq to show that the Set3C acts as a transcriptional co-factor of metabolic and morphogenesis-related genes in C. albicans. Binding of the Set3C correlates with gene expression during fungal morphogenesis; yet, surprisingly, deletion of SET3 leaves the steady-state expression level of most genes unchanged, both during exponential yeast-phase growth and during the yeast-filament transition. Fine temporal resolution of transcription in cells undergoing this transition revealed that the Set3C modulates transient expression changes of key morphogenesis-related genes. These include a transcription factor cluster comprising of NRG1, EFG1, BRG1, and TEC1, which form a regulatory circuit controlling hyphal differentiation. Set3C appears to restrict the factors by modulating their transcription kinetics, and the hyperfilamentous phenotype of SET3-deficient cells can be reverted by mutating the circuit factors. These results indicate that the chromatin status at coding regions represents a dynamic platform influencing transcription kinetics. Moreover, we suggest that transcription at the coding sequence can be transiently decoupled from potentially conflicting promoter information in dynamic environments.
Charcot-Marie-Tooth type 2B (CMT2B) is a peripheral ulcero-mutilating neuropathy caused by four missense mutations in the rab7a gene. CMT2B is clinically characterized by prominent sensory loss, distal muscle weakness leading to muscle atrophy, high frequency of foot ulcers and infections that often results in toe amputations. RAB7A is a ubiquitous small GTPase, which controls transport to late endocytic compartments. Although the biochemical and functional properties of disease-causing RAB7A mutant proteins have been investigated, it is not yet clear how the disease originates. To understand how mutations in a ubiquitous protein specifically affect peripheral neurons, we performed a two-hybrid screen using a dorsal root ganglia cDNA library with the purpose of identifying RAB7A interactors specific for these cells. We identified peripherin, an intermediate filament protein expressed primarily in peripheral neurons, as a putative RAB7A interacting protein. The interaction was confirmed by co-immunoprecipitation and pull-down experiments, and established that the interaction is direct using recombinant proteins. Silencing or overexpression of wild type RAB7A changed the soluble/insoluble rate of peripherin indicating that RAB7A is important for peripherin organization and function. In addition, disease-causing RAB7A mutant proteins bind more strongly to peripherin and their expression causes a significant increase in the amount of soluble peripherin. Since peripherin plays a role not only in neurite outgrowth during development but also in axonal regeneration after injury, these data suggest that the altered interaction between disease-causing RAB7A mutants and peripherin could play an important role in CMT2B neuropathy.
Ubiquitination plays an important role in the DNA damage response. We identified a novel interaction of the E3 ubiquitin ligase RNF8 with Nbs1, a key regulator of DNA double-strand break (DSB) repair. We found that Nbs1 is ubiquitinated both before and after DNA damage and is a direct ubiquitination substrate of RNF8. We also identified key residues on Nbs1 that are ubiquitinated by RNF8. By using laser microirradiation and live-cell imaging, we observed that RNF8 and its ubiquitination activity are important for promoting optimal binding of Nbs1 to DSB-containing chromatin. We also demonstrated that RNF8-mediated ubiquitination of Nbs1 contributes to the efficient and stable binding of Nbs1 to DSBs and is important for HR-mediated DSB repair. Taken together, these studies suggest that Nbs1 is one important target of RNF8 to regulate DNA DSB repair.
Budding yeast centromeres are sequence-defined point centromeres and are, unlike in many other organisms, not embedded in heterochromatin. Here we show that Fun30, a poorly understood SWI/SNF-like chromatin remodeling factor conserved in humans, promotes point centromere function through the formation of correct chromatin architecture at centromeres. Our determination of the genome-wide binding and nucleosome positioning properties of Fun30 shows that this enzyme is consistently enriched over centromeres and that a majority of CENs show Fun30-dependent changes in flanking nucleosome position and/or CEN core micrococcal nuclease accessibility. Fun30 deletion leads to defects in histone variant Htz1 occupancy genome-wide, including at and around most centromeres. FUN30 genetically interacts with CSE4, coding for the centromere-specific variant of histone H3, and counteracts the detrimental effect of transcription through centromeres on chromosome segregation and suppresses transcriptional noise over centromere CEN3. Previous work has shown a requirement for fission yeast and mammalian homologs of Fun30 in heterochromatin assembly. As centromeres in budding yeast are not embedded in heterochromatin, our findings indicate a direct role of Fun30 in centromere chromatin by promoting correct chromatin architecture.
The Mre11-Rad50-Nbs1 (MRN) complex plays critical roles in checkpoint activation and double-stranded break (DSB) repair. The Rad50 zinc hook domain mediates zinc-dependent intercomplex associations of MRN, which is important for DNA tethering. Studies in yeast suggest that the Rad50 zinc hook domain is essential for MRN functions, but its role in mammalian cells is not clear. We demonstrated that the human Rad50 hook mutants are severely defective in various DNA damage responses including ATM (Ataxia telangiectasia mutated) activation, homologous recombination, sensitivity to IR, and activation of the ATR pathway. By using live cell imaging, we observed that the Rad50 hook mutants fail to be recruited to chromosomal DSBs, suggesting a novel mechanism underlying the severe defects observed for the Rad50 hook mutants. In vitro analysis showed that Zn(2+) promotes wild type but not the hook mutant of MR to bind double-stranded DNA. In vivo, the Rad50 hook mutants are defective in being recruited to chromosomal DSBs in both H2AX-proficient and -deficient cells, suggesting that the Rad50 hook mutants are impaired in direct binding to chromosomal DSB ends. We propose that the Rad50 zinc hook domain is important for the initial binding of MRN to DSBs, leading to ATM activation to phosphorylate H2AX, which recruits more MRN to the DSB-flanking chromosomal regions. Our studies reveal a critical role for the Rad50 zinc hook domain in establishing and maintaining MRN recruitment to chromosomal DSBs and suggest an important mechanism of how the Rad50 zinc hook domain contributes to DNA repair and checkpoint activation.
Dyskinesia, a motor complication caused by prolonged administration of the antiparkinsonian drug l-3,4-dihydroxyphenylalanine (l-DOPA), is accompanied by activation of cAMP signaling and hyperphosphorylation of the dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32). Here, we show that the abnormal phosphorylation of DARPP-32 occurs specifically in medium spiny neurons (MSNs) expressing dopamine D1 receptors (D1R). Using mice in which DARPP-32 is selectively deleted in D1R-expressing MSNs, we demonstrate that this protein is required for l-DOPA-induced activation of the extracellular signal-regulated protein kinases 1 and 2 and the mammalian target of rapamycin complex 1 (mTORC1) pathways, which are implicated in dyskinesia. We also show that mutation of the phosphorylation site for cAMP-dependent protein kinase on DARPP-32 attenuates l-DOPA-induced dyskinesia and reduces the concomitant activations of ERK and mTORC1 signaling. These studies demonstrate that, in D1R-expressing MSNs, l-DOPA-induced activation of ERK and mTORC1 requires DARPP-32 and indicates the importance of the cAMP/DARPP-32 signaling cascade in dyskinesia.
CtIP (CtBP-interacting protein) associates with BRCA1 and the Mre11-Rad50-Nbs1 (MRN) complex and plays an essential role in homologous recombination (HR)-mediated DNA double-stranded break (DSB) repair. It has been described that CtIP forms dimers in mammalian cells, but the biological significance is not clear. In this study, we identified a conserved motif in the N terminus of CtIP, which is required for dimer formation. We further showed that CtIP mutants impaired in forming dimers are strongly defective in HR, end resection, and activation of the ataxia telangiectasia and Rad3-related pathway, without notable change of CtIP interactions with BRCA1 or Nbs1. In addition to HR, CtIP dimerization is also required for microhomology-mediated end joining. Live cell imaging of enhanced GFP-tagged CtIP demonstrates that the CtIP dimerization mutant fails to be localized to DSBs, whereas placing a heterologous dimerization motif to the dimerization mutant restores CtIP recruitment to DSBs. These studies suggest that CtIP dimer formation is essential for its recruitment to DSBs on chromatin upon DNA damage. Furthermore, DNA damage-induced phosphorylation of CtIP is significantly reduced in the CtIP dimerization mutants. Therefore, in addition to the C-terminal conserved domains critical for CtIP function, the dimerization motif on the N terminus of CtIP is also conserved and essential for its function in DNA damage responses. The severe repair defects of CtIP dimerization mutants are likely due to the failure in localization to chromosomal DSBs upon DNA damage.
Important cellular processes are regulated by poly(ADP-ribosyl)ation. This protein modification is catalyzed mainly by nuclear poly(ADP-ribose) polymerase (PARP) 1 in response to DNA damage. Cytosolic PARP isoforms have been described, whereas the presence of poly(ADP-ribose) (PAR) metabolism in mitochondria is controversial. PAR is degraded by poly(ADP-ribose) glycohydrolase (PARG). Recently, ADP-ribosylhydrolase 3 (ARH3) was also shown to catalyze PAR-degradation in vitro. PARG is encoded by a single, essential gene. One nuclear and three cytosolic isoforms result from alternative splicing. The presence and origin of a mitochondrial PARG is still unresolved. We establish here the genetic background of a human mitochondrial PARG isoform and investigate the molecular basis for mitochondrial poly(ADP-ribose) degradation. In common with a cytosolic 60-kDa human PARG isoform, the mitochondrial protein did not catalyze PAR degradation because of the absence of exon 5-encoded residues. In mice, we identified a transcript encoding an inactive cytosolic 52-kDa PARG lacking the mitochondrial targeting sequence and a substantial portion of exon 5. Thus, mammalian PARG genes encode isoforms that do not catalyze PAR degradation. On the other hand, embryonic fibroblasts from ARH3(-/-) mice lack most of the mitochondrial PAR degrading activity detected in wild-type cells, demonstrating a potential involvement of ARH3 in PAR metabolism.
During mitosis in budding yeast, cortically anchored dynein generates pulling forces on astral microtubules to position the mitotic spindle across the mother-bud neck. The attachment molecule Num1 is required for dynein anchoring at the cell membrane, but how Num1 assembles into stationary cortical patches and interacts with dynein is unknown. We show that an N-terminal Bin/Amphiphysin/Rvs (BAR)-like domain in Num1 mediates the assembly of morphologically distinct patches and its interaction with dynein for spindle translocation into the bud. We name this domain patch assembly domain (PA; residues 1-303), as it was both necessary and sufficient for the formation of functional dynein-anchoring patches when it was attached to a pleckstrin homology domain or a CAAX motif. Distinct point mutations targeting the predicted BAR-like PA domain differentially disrupted patch assembly, dynein anchoring, and mitochondrial attachment functions of Num1. We also show that the PA domain is an elongated dimer and discuss the mechanism by which it drives patch assembly.
Accumulation of the neurotoxic β-amyloid (Aβ) peptide in the brain is central to the pathogenesis of Alzheimer disease. Aβ is derived from the β-amyloid precursor protein (APP) through sequential cleavages by β- and γ-secretases, and the production of Aβ is greatly affected by the subcellular localization of these factors. CUTA, the mammalian CutA divalent cation tolerance homolog (E. coli), has been proposed to mediate acetylcholinesterase activity and copper homeostasis, which are important in Alzheimer disease pathology. However, the exact function of CUTA remains largely unclear. Here we show that human CUTA has several variants that differ in their N-terminal length and are separated as heavy (H) and light (L) components. The H component has the longest N terminus and is membrane-associated, whereas the L component is N-terminally truncated at various sites and localized in the cytosol. Importantly, we demonstrate that the H component of CUTA interacts through its N terminus with the transmembrane domain of β-site APP cleaving enzyme 1 (BACE1), the putative β-secretase, mainly in the Golgi/trans-Golgi network. Overexpression and RNA interference knockdown of CUTA can reduce and increase BACE1-mediated APP processing/Aβ secretion, respectively. RNA interference of CUTA decelerates intracellular trafficking of BACE1 from the Golgi/trans-Golgi network to the cell surface and reduces the steady-state level of cell surface BACE1. Our results identify the H component of CUTA as a novel BACE1-interacting protein that mediates the intracellular trafficking of BACE1 and the processing of APP to Aβ.
Nucleostemin (NS), a nucleolar GTPase, is highly expressed in stem/progenitor cells and in most cancer cells. However, little is known about the regulation of its expression. Here, we identify the NS gene as a novel direct transcriptional target of the c-Myc oncoprotein. We show that Myc overexpression enhances NS transcription in cultured cells and in pre-neoplastic B cells from Eμ-myc transgenic mice. Consistent with NS being downstream of Myc, NS expression parallels that of Myc in a large panel of human cancer cell lines. Using chromatin immunoprecipitation we show that c-Myc binds to a well-conserved E-box in the NS promoter. Critically, we show NS haploinsufficiency profoundly delays Myc-induced cancer formation in vivo. NS+/-Eμ-myc transgenic mice have much slower rates of B-cell lymphoma development, with life spans twice that of their wild-type littermates. Moreover, we demonstrate that NS is essential for the proliferation of Myc-overexpressing cells in cultured cells and in vivo: impaired lymphoma development was associated with a drastic decrease of c-Myc-induced proliferation of pre-tumoural B cells. Finally, we provide evidence that in cell culture NS controls cell proliferation independently of p53 and that NS haploinsufficiency significantly delays lymphomagenesis in p53-deficient mice. Together these data indicate that NS functions downstream of Myc as a rate-limiting regulator of cell proliferation and transformation, independently from its putative role within the p53 pathway. Targeting NS is therefore expected to compromise early tumour development irrespectively of the p53 status.
Sorting nexin 27 (SNX27) is a 62-kDa protein localized to early endosomes and known to regulate the intracellular trafficking of ion channels and receptors. In addition to a PX domain, SNX27 is the only sorting family member that contains a PDZ domain. To identify novel SNX27-PDZ binding partners, we performed a proteomic screen in mouse principal kidney cortical collecting duct cells using a GST-SNX27 fusion construct as bait. We found that β-Pix (p21-activated kinase-interactive exchange factor), a guanine nucleotide exchange factor for the Rho family of small GTPases known to regulate cell motility directly interacted with SNX27. The association of β-Pix and SNX27 is specific for β-Pix isoforms terminating in the type-1 PDZ binding motif (ETNL). In the same screen we also identified Git1/2 as a potential SNX27 interacting protein. The interaction between SNX27 and Git1/2 is indirect and mediated by β-Pix. Furthermore, we show recruitment of the β-Pix·Git complex to endosomal sites in a SNX27-dependent manner. Finally, migration assays revealed that depletion of SNX27 from HeLa and mouse principal kidney cortical collecting duct cells significantly decreases cell motility. We propose a model by which SNX27 regulates trafficking of β-Pix to focal adhesions and thereby influences cell motility.
Podosomes are dynamic actin-rich adhesion plasma membrane microdomains endowed with extracellular matrix-degrading activities. In aortic endothelial cells, podosomes are induced by transforming growth factor β (TGF-β), but how this occurs is largely unknown. It is thought that, in endothelial cells, podosomes play a role in vessel remodeling and/or in breaching anatomical barriers. We demonstrate here that, in bovine aortic endothelial cells, that the Cdc42-specific guanine exchange factor (GEF) Fgd1 is expressed and regulated by TGF-β to induce Cdc42-dependent podosome assembly. Within 15 min of TGF-β stimulation, Fgd1, but none of the other tested Cdc42 GEFs, undergoes tyrosine phosphorylation, associates with Cdc42, and translocates to the subcortical cytoskeleton via a cortactin-dependent mechanism. Small interfering RNA-mediated Fgd1 knockdown inhibits TGF-β-induced Cdc42 activation. Fgd1 depletion also reduces podosome formation and associated matrix degradation and these defects are rescued by reexpression of Fgd1. Although overexpression of Fgd1 does not promote podosome formation per se, it enhances TGF-β-induced matrix degradation. Our results identify Fgd1 as a TGF-β-regulated GEF and, as such, the first GEF to be involved in the process of cytokine-induced podosome formation. Our findings reveal the involvement of Fgd1 in endothelial cell biology and open up new avenues to study its role in vascular pathophysiology.
In Drosophila, Piwi proteins associate with Piwi-interacting RNAs (piRNAs) and protect the germline genome by silencing mobile genetic elements. This defense system acts in germline and gonadal somatic tissue to preserve germline development. Genetic control for these silencing pathways varies greatly between tissues of the gonad. Here, we identified Vreteno (Vret), a novel gonad-specific protein essential for germline development. Vret is required for piRNA-based transposon regulation in both germline and somatic gonadal tissues. We show that Vret, which contains Tudor domains, associates physically with Piwi and Aubergine (Aub), stabilizing these proteins via a gonad-specific mechanism that is absent in other fly tissues. In the absence of vret, Piwi-bound piRNAs are lost without changes in piRNA precursor transcript production, supporting a role for Vret in primary piRNA biogenesis. In the germline, piRNAs can engage in an Aub- and Argonaute 3 (AGO3)-dependent amplification in the absence of Vret, suggesting that Vret function can distinguish between primary piRNAs loaded into Piwi-Aub complexes and piRNAs engaged in the amplification cycle. We propose that Vret plays an essential role in transposon regulation at an early stage of primary piRNA processing.
MCM proteins are components of a DNA helicase that plays an essential role in DNA replication and cell proliferation. However, MCM proteins are present in excess relative to origins of replication, suggesting they may serve other functions. Decreased proliferation is a fundamental physiological response to hypoxia in many cell types, and hypoxia-inducible factor 1 (HIF-1) has been implicated in this process. Here, we demonstrate that multiple MCM proteins bind directly to the HIF-1α subunit and synergistically inhibit HIF-1 transcriptional activity via distinct O(2)-dependent mechanisms. MCM3 inhibits transactivation domain function, whereas MCM7 enhances HIF-1α ubiquitination and proteasomal degradation. HIF-1 activity decreases when quiescent cells re-enter the cell cycle, and this effect is MCM dependent. Exposure to hypoxia leads to MCM2-7 downregulation in diverse cell types. These studies reveal a function of MCM proteins apart from their DNA helicase activity and establish a direct link between HIF-1 and the cell-cycle machinery.
The intensity of gene transcription is generally reflected by the level of RNA polymerase II (RNAPII) recruitment to the gene. However, genome-wide studies of polymerase occupancy indicate that RNAPII distribution varies among genes. In some loci more polymerases are found in the 5' region, whereas in other loci, in the 3' region of the gene. We studied the distribution of elongating RNAPII complexes at highly transcribed GAL-VPS13 locus in Saccharomyces cerevisiae and found that in the cell population the amount of polymerases gradually decreased toward the 3' end of the gene. However, the conventional chromatin immunoprecipitation assay averages the signal from the cell population, and no data on single cell level can be gathered. To study the spacing of elongating polymerases on single chromosomes, we used a sequential chromatin immunoprecipitation assay for the detection of multiple RNAPII complexes on the same DNA fragment. Our results demonstrate uniform distribution of elongating polymerases throughout all regions of the GAL-VPS13 gene.
Insulin-degrading enzyme (IDE) is a Zn(2+) metalloprotease with a characteristic inverted catalytic motif. IDE is ubiquitously expressed and degrades peptide substrates including insulin, endorphin, and the amyloid-β peptide. Although IDE is mainly expressed in the cytosol, it can also be found on the cell surface and in secreted form in extracellular fluids. As IDE lacks a characteristic signal sequence that targets the protein to the classical secretory pathway, release of the enzyme involves non-conventional mechanisms. However, functional domains of IDE involved in its secretion remain elusive. By bioinformatical, biochemical, and cell biological methods, we identified a novel amino acid motif ((853)EKPPHY(858)) close to the C terminus of IDE and characterized its function in the non-conventional secretion of the protein. Because of its close homology to an amino acid sequence found in bacterial proteins belonging to the SlyX family, we propose to call it the SlyX motif. Mutagenesis revealed that deletion of this motif strongly decreased the release of IDE, whereas deletion of a potential microbody-targeting signal at the extreme C terminus had little effect on secretion. The combined data indicate that the non-conventional secretion of IDE is regulated by the newly identified SlyX motif.
Yeast cells lacking Ctf18, the major subunit of an alternative Replication Factor C complex, have multiple problems with genome stability. To understand the in vivo function of the Ctf18 complex, we analyzed chromatin composition in a ctf18Δ mutant using the quantitative proteomic technique of stable isotope labeling by amino acids in cell culture. Three hundred and seven of the 491 reported chromosomal proteins were quantitated. The most marked abnormalities occurred when cells were challenged with the replication inhibitor hydroxyurea. Compared with wild type, hydroxyurea-treated ctf18Δ cells exhibited increased chromatin association of replisome progression complex components including Cdc45, Ctf4, and GINS complex subunits, the polymerase processivity clamp PCNA and the single-stranded DNA-binding complex RPA. Chromatin composition abnormalities observed in ctf18Δ cells were very similar to those of an mrc1Δ mutant, which is defective in the activating the Rad53 checkpoint kinase in response to DNA replication stress. We found that ctf18Δ cells are also defective in Rad53 activation, revealing that the Ctf18 complex is required for engagement of the DNA replication checkpoint. Inappropriate initiation of replication at late origins, because of loss of the checkpoint, probably causes the elevated level of chromatin-bound replisome proteins in the ctf18Δ mutant. The role of Ctf18 in checkpoint activation is not shared by all Replication Factor C-like complexes, because proteomic analysis revealed that cells lacking Elg1 (the major subunit of a different Replication Factor C-like complex) display a different spectrum of chromatin abnormalities. Identification of Ctf18 as a checkpoint protein highlights the usefulness of chromatin proteomic analysis for understanding the in vivo function of proteins that mediate chromatin transactions.
Deregulation of the ubiquitin-protein ligase E6AP contributes to the development of the Angelman syndrome and to cervical carcinogenesis suggesting that the activity of E6AP needs to be under tight control. However, how E6AP activity is regulated at the post-translational level under non-pathologic conditions is poorly understood. In this study, we report that the giant protein HERC2, which is like E6AP a member of the HECT family of ubiquitin-protein ligases, binds to E6AP. The interaction is mediated by the RCC1-like domain 2 of HERC2 and a region spanning amino acid residues 150-200 of E6AP. Furthermore, we provide evidence that HERC2 stimulates the ubiquitin-protein ligase activity of E6AP in vitro and within cells and that this stimulatory effect does not depend on the ubiquitin-protein ligase activity of HERC2. Thus, the data obtained indicate that HERC2 acts as a regulator of E6AP.
Here we describe the isolation, characterisation and ex-vivo expansion of human epidermal neural crest stem cells (hEPI-NCSC) and we provide protocols for their directed differentiation into osteocytes and melanocytes. hEPI-NCSC are neural crest-derived multipotent stem cells that persist into adulthood in the bulge of hair follicles. Multipotency and self-renewal were determined by in vitro clonal analyses. hEPI-NCSC generate all major neural crest derivatives, including bone/cartilage cells, neurons, Schwann cells, myofibroblasts and melanocytes. Furthermore, hEPI-NCSC express additional neural crest stem cell markers and global stem cell genes. To variable degrees and in a donor-dependent manner, hEPI-NCSC express the six essential pluripotency genes C-MYC, KLF4, SOX2, LIN28, OCT-4/POU5F1 and NANOG. hEPI-NCSC can be expanded ex vivo into millions of stem cells that remain mulitpotent and continue to express stem cell genes. The novelty of hEPI-NCSC lies in the combination of their highly desirable traits. hEPI-NCSC are embryonic remnants in a postnatal location, the bulge of hair follicles. Therefore they are readily accessible in the hairy skin by minimal invasive procedure. hEPI-NCSC are multipotent somatic stem cells that can be isolated reproducibly and with high yield. By taking advantage of their migratory ability, hEPI-NCSC can be isolated as a highly pure population of stem cells. hEPI-NCSC can undergo robust ex vivo expansion and directed differentiation. As somatic stem cells, hEPI-NCSC are conducive to autologous transplantation, which avoids graft rejection. Together, these traits make hEPI-NCSC novel and attractive candidates for future cell-based therapies and regenerative medicine.
FACT complex is involved in elongation and ensures fidelity in the initiation step of transcription by RNA polymerase (pol) II. Histone variant H2A.Z is found in nucleosomes at the 5'-end of many genes. We report here H2A.Z-chaperone activity of the yeast FACT complex on the short, nucleosome-free, non-coding, pol III-transcribed yeast tRNA genes. On a prototype gene, yeast SUP4, chromatin remodeler RSC and FACT regulate its transcription through novel mechanisms, wherein the two gene-flanking nucleosomes containing H2A.Z, play different roles. Nhp6, which ensures transcription fidelity and helps load yFACT onto the gene flanking nucleosomes, has inhibitory role. RSC maintains a nucleosome abutting the gene terminator downstream, which results in reduced transcription rate in active state while H2A.Z probably helps RSC in keeping the gene nucleosome-free and serves as stress-sensor. All these factors maintain an epigenetic state which allows the gene to return quickly from repressed to active state and tones down the expression from the active SUP4 gene, required probably to maintain the balance in cellular tRNA pool.
MicroRNAs (miRNAs) are small regulatory RNAs targeting multiple effectors of cell homeostasis and development, whose malfunctions are associated with major pathologies such as cancer. Herein we show that GAM/ZFp/ZNF512B works within an intricate gene regulatory network involving cell-cycle regulators, TGFβ effectors and oncogenic miRNAs of the miR-17-92 cluster. Thus, GAM impairs the transcriptional activation of the miR-17-92 promoter by c-Myc, downregulates miR-17-92 miRNAs differentially, and limits the activation of genes responsive to TGFβ canonical pathway. In contrast, TGFβ decreases GAM transcripts levels while differentially upregulating miR-17-92 miRNAs. In turn, miR-17, miR-20a and miR-92a-1 target GAM transcripts, thus establishing a feedback autoregulatory loop. GAM transcripts are also targeted by miRNAs of the let-7 family. GAM downregulates Drosha, the main effector of miRNA maturation in the nucleus, and interacts with it in a RNA-dependent manner. Finally, GAM modulates the levels of E2F1 and Ras, and increases apoptosis while reducing cell proliferation. We propose that GAM represents a new kind of vertebrate regulator aimed at balancing the opposite effects of regulators of cell homeostasis by increasing the robustness of gene circuitries controlling cell proliferation, differentiation and development.
Bipolar spindle formation is essential for faithful chromosome segregation at mitosis. Because centrosomes define spindle poles, abnormal number and structural organization of centrosomes can lead to loss of spindle bipolarity and genetic integrity. ASAP (aster-associated protein or MAP9) is a centrosome- and spindle-associated protein, the deregulation of which induces severe mitotic defects. Its phosphorylation by Aurora A is required for spindle assembly and mitosis progression. Here, we show that ASAP is localized to the spindle poles by Polo-like kinase 1 (Plk1) (a mitotic kinase that plays an essential role in centrosome regulation and mitotic spindle assembly) through the γ-TuRC-dependent pathway. We also demonstrate that ASAP is a novel substrate of Plk1 phosphorylation and have identified serine 289 as the major phosphorylation site by Plk1 in vivo. ASAP phosphorylated on serine 289 is localized to centrosomes during mitosis, but this phosphorylation is not required for its Plk1-dependent localization at the spindle poles. We show that phosphorylated ASAP on serine 289 contributes to spindle pole stability in a microtubule-dependent manner. These data reveal a novel function of ASAP in centrosome integrity. Our results highlight dual ASAP regulation by Plk1 and further confirm the importance of ASAP for spindle pole organization, bipolar spindle assembly, and mitosis.
Polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome (PMSE) is a rare human autosomal-recessive disorder characterized by abnormal brain development, cognitive disability, and intractable epilepsy. It is caused by homozygous deletions of STE20-related kinase adaptor alpha (STRADA). The underlying pathogenic mechanisms of PMSE and the role of STRADA in cortical development remain unknown. Here, we found that a human PMSE brain exhibits cytomegaly, neuronal heterotopia, and aberrant activation of mammalian target of rapamycin complex 1 (mTORC1) signaling. STRADalpha normally binds and exports the protein kinase LKB1 out of the nucleus, leading to suppression of the mTORC1 pathway. We found that neurons in human PMSE cortex exhibited abnormal nuclear localization of LKB1. To investigate this further, we modeled PMSE in mouse neural progenitor cells (mNPCs) in vitro and in developing mouse cortex in vivo by knocking down STRADalpha expression. STRADalpha-deficient mNPCs were cytomegalic and showed aberrant rapamycin-dependent activation of mTORC1 in association with abnormal nuclear localization of LKB1. Consistent with the observations in human PMSE brain, knockdown of STRADalpha in vivo resulted in cortical malformation, enhanced mTORC1 activation, and abnormal nuclear localization of LKB1. Thus, we suggest that the aberrant nuclear accumulation of LKB1 caused by STRADalpha deficiency contributes to hyperactivation of mTORC1 signaling and disruption of neuronal lamination during corticogenesis, and thereby the neurological features associated with PMSE.
Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) and PARK2/Parkin mutations cause autosomal recessive forms of Parkinson's disease. Upon a loss of mitochondrial membrane potential (DeltaPsi(m)) in human cells, cytosolic Parkin has been reported to be recruited to mitochondria, which is followed by a stimulation of mitochondrial autophagy. Here, we show that the relocation of Parkin to mitochondria induced by a collapse of DeltaPsi(m) relies on PINK1 expression and that overexpression of WT but not of mutated PINK1 causes Parkin translocation to mitochondria, even in cells with normal DeltaPsi(m). We also show that once at the mitochondria, Parkin is in close proximity to PINK1, but we find no evidence that Parkin catalyzes PINK1 ubiquitination or that PINK1 phosphorylates Parkin. However, co-overexpression of Parkin and PINK1 collapses the normal tubular mitochondrial network into mitochondrial aggregates and/or large perinuclear clusters, many of which are surrounded by autophagic vacuoles. Our results suggest that Parkin, together with PINK1, modulates mitochondrial trafficking, especially to the perinuclear region, a subcellular area associated with autophagy. Thus by impairing this process, mutations in either Parkin or PINK1 may alter mitochondrial turnover which, in turn, may cause the accumulation of defective mitochondria and, ultimately, neurodegeneration in Parkinson's disease.
Spongiform neurodegeneration is characterized by the appearance of vacuoles throughout the central nervous system. It has many potential causes, but the underlying cellular mechanisms are not well understood. Mice lacking the E3 ubiquitin ligase Mahogunin Ring Finger-1 (MGRN1) develop age-dependent spongiform encephalopathy. We identified an interaction between a "PSAP" motif in MGRN1 and the ubiquitin E2 variant (UEV) domain of TSG101, a component of the endosomal sorting complex required for transport I (ESCRT-I), and demonstrate that MGRN1 multimonoubiquitinates TSG101. We examined the in vivo consequences of loss of MGRN1 on TSG101 expression and function in the mouse brain. The pattern of TSG101 ubiquitination differed in the brains of wild-type mice and Mgrn1 null mutant mice: at 1 month of age, null mutant mice had less ubiquitinated TSG101, while in adults, mutant mice had more ubiquitinated, insoluble TSG101 than wild-type mice. There was an associated increase in epidermal growth factor receptor (EGFR) levels in mutant brains. These results suggest that loss of MGRN1 promotes ubiquitination of TSG101 by other E3s and may prevent its disassociation from endosomal membranes or cause it to form insoluble aggregates. Our data implicate loss of normal TSG101 function in endo-lysosomal trafficking in the pathogenesis of spongiform neurodegeneration in Mgrn1 null mutant mice.
A growing body of experimental and clinical studies supports a strong association between psychological stress and cardiovascular disease. An important endogenous cardioprotective role in heart physiology has been attributed to corticotropin-releasing factor receptor type 2beta (CRFR2beta). Here, we report the isolation of cDNA from mouse (m) heart encoding a novel CRFR2beta splice variant. Translation of this insertion variant (iv)-mCRFR2beta isoform produces a 421-aa protein that includes a unique C-terminal cytoplasmic tail. Our functional analysis and cellular localization studies demonstrated that when coexpressed with wild-type mCRFR2beta, iv-mCRFR2beta significantly inhibited the wild-type mCRFR2beta membrane expression and its functional signaling by ER-Golgi complex retention, suggesting a dose-dependent dominant negative effect. Interestingly, mice exposed to a 4-wk paradigm of chronic variable stress, a model of chronic psychological stress in humans, presented significantly lower levels of mCRFR2beta and higher levels of iv-mCRFR2beta mRNA expression in their hearts, compared to nonstressed control mice. The dominant-negative effect of iv-mCRFR2beta and its up-regulation by psychological stress suggest a new form of regulation of the mCRFR2beta cardioprotective effect and a potential role for this novel isoform in stress-induced heart disease.
Huntington's disease (HD) is a fatal neurodegenerative disease caused by mutant huntingtin (htt) protein, and there are currently no effective treatments. Recently, we and others demonstrated that silencing mutant htt via RNA interference (RNAi) provides therapeutic benefit in HD mice. We have since found that silencing wild-type htt in adult mouse striatum is tolerated for at least 4 months. However, given the role of htt in various cellular processes, it remains unknown whether nonallele-specific silencing of both wild-type and mutant htt is a viable therapeutic strategy for HD. Here, we tested whether cosilencing wild-type and mutant htt provides therapeutic benefit and is tolerable in HD mice. After treatment, HD mice showed significant reductions in wild-type and mutant htt, and demonstrated improved motor coordination and survival. We performed transcriptional profiling to evaluate the effects of reducing wild-type htt in adult mouse striatum. We identified gene expression changes that are concordant with previously described roles for htt in various cellular processes. Also, several abnormally expressed transcripts associated with early-stage HD were differentially expressed in our studies, but intriguingly, those involved in neuronal function changed in opposing directions. Together, these encouraging and surprising findings support further testing of nonallele-specific RNAi therapeutics for HD.
RhoBTB proteins are atypical members of the Rho family of small GTPases. Two of the three RhoBTB proteins, RhoBTB1 and RhoBTB2, have been proposed as tumor suppressors and might function as adaptors of Cul3-dependent ubiquitin ligase complexes. Using yeast two-hybrid analysis and co-immunoprecipitation we show that all three RhoBTB proteins interact with Cul3. The interaction requires the N-terminal region of Cul3 and the first BTB domain of RhoBTB. RhoBTB3, the only RhoBTB with a prenylation motif, associates with vesicles that are frequently found in the vicinity of microtubules, suggesting a participation in some aspects of vesicle trafficking. We also show that RhoBTB2 and RhoBTB3 are capable of homo and heterodimerizing through the BTB domain region. The GTPase domain, which does not bind GTP, is able to interact with the BTB domain region, thus preventing proteasomal degradation of RhoBTB. This fits into a model in which an intramolecular interaction maintains RhoBTB in an inactive state, preventing the formation or the functionality of Cul3-dependent complexes. We also report a significantly decreased expression of RHOBTB and CUL3 genes in kidney and breast tumor samples and a very good correlation in the expression changes between RHOBTB and CUL3 that suggests that these genes are subject to a common inactivation mechanism in tumors.
Enhancer of zeste homolog 2 (EZH2) is a critical component of the polycomb-repressive complex 2 (PRC2), which is involved in gene silencing and histone H3 lysine 27 methylation. EZH2 has a master regulatory function in controlling such processes as stem cell differentiation, cell proliferation, early embryogenesis and X chromosome inactivation. Although benign epithelial cells express very low levels of EZH2, increased levels of EZH2 have been observed in aggressive solid tumors such as those of the prostate, breast and bladder. The mechanism by which EZH2 mediates tumor aggressiveness is unclear. Here, we demonstrate that EZH2 mediates transcriptional silencing of the tumor suppressor gene E-cadherin by trimethylation of H3 lysine 27. Histone deacetylase inhibitors can prevent EZH2-mediated repression of E-cadherin and attenuate cell invasion, suggesting a possible mechanism that may be useful for the development of therapeutic treatments. Taken together, these observations provide a novel mechanism of E-cadherin regulation and establish a functional link between dysregulation of EZH2 and repression of E-cadherin during cancer progression.
Palmitoylation is the post-translational addition of a palmitate moiety to a cysteine residue through a covalent thioester bond. The addition and removal of this modification is controlled by both palmitoyl acyl-transferases and thioesterases. Using bioinformatic analysis, we identified 22 DHHC family palmitoyl acyl-transferase homologs in the Drosophila genome. We used in situ hybridization,RT-PCR, and published FlyAtlas microarray data to characterize the expression patterns of all 22 fly homologs. Our results indicate that all are expressed genes, but several, including CG1407, CG4676, CG5620, CG6017/dHIP14, CG6618, CG6627 and CG17257 appear to be enriched in neural tissues suggesting that they are important for neural function. Furthermore, we have found that several may be expressed in a sex-specific manner with adult male specific expression of CG4483 and CG17195. Using tagged versions of the DHHC genes, we demonstrate that fly DHHC proteins are primarily located in either the Golgi Apparatus or Endoplasmic Reticulum in S2 cells, except for CG1407, which was found on the plasma membrane. We also characterized the subcellular localization and expression of the three known thioesterases: Palmitoyl-protein Thioesterase 1 (Ppt1), Palmitoyl-protein Thioesterase 2 (Ppt2)and Acyl-protein Thioesterase 1 (APT1). Our results indicate that Ppt1 and Ppt2 are the major lysosomal thioesterases while APT1 is the likely cytoplasmic thioesterase. Finally, in vivo rescue experiments show that Ppt2 expression cannot rescue the neural inclusion phenotypes associated with loss of Ppt1, further supporting distinct functions and substrates for these two thioesterases. These results will serve as the basis for a more complete understanding of the protein palmitoylome's normal cellular functions in the fly and will lead to further insights into the molecular etiology of diseases associated with the mis-regulation of palmitoylation.
Despite the importance of airspace integrity in vertebrate gas exchange, the molecular pathways that instruct distal lung formation are poorly understood. Recently, we found that fibrillin-1 deficiency in mice impairs alveolar formation and recapitulates the pulmonary features of human Marfan syndrome. To further elucidate effectors involved in distal lung formation, we performed expression profiling analysis comparing the fibrillin-1-deficient and wild-type developing lung. NeuroD, a basic helix-loop-helix transcription factor, fulfilled the expression criteria for a candidate mediator of distal lung development. We investigated its role in murine lung development using genetically targeted NeuroD-deficient mice. We found that NeuroD deficiency results in both impaired alveolar septation and altered morphology of the pulmonary neuroendocrine cells. NeuroD-deficient mice had enlarged alveoli associated with reduced epithelial proliferation in the airway and airspace compartments during development. Additionally, the neuroendocrine compartment in these mice manifested an increased number of neuroepithelial bodies but a reduced number of solitary pulmonary neuroendocrine cells in the neonatal lung. Overexpression of NeuroD in a murine lung epithelial cell line conferred a neuroendocrine phenotype characterized by the induction of neuroendocrine markers as well as increased proliferation. These results support an unanticipated role for NeuroD in the regulation of pulmonary neuroendocrine and alveolar morphogenesis and suggest an intimate connection between the neuroendocrine compartment and distal lung development.
Sm-like (Lsm) proteins are ubiquitous, multifunctional proteins that are involved in the processing and/or turnover of many RNAs. In eukaryotes, a hetero-heptameric complex of seven Lsm proteins (Lsm2-8) affects the processing of small stable RNAs and pre-mRNAs in the nucleus, whereas a different hetero-heptameric complex of Lsm proteins (Lsm1-7) promotes mRNA decapping and decay in the cytoplasm. These two complexes have six constituent proteins in common, yet localize to separate cellular compartments and perform apparently disparate functions. Little is known about the biogenesis of the Lsm complexes, or how they are recruited to different cellular compartments. We show that, in yeast, the nuclear accumulation of Lsm proteins depends on complex formation and that the Lsm8p subunit plays a crucial role. The nuclear localization of Lsm8p is itself most strongly influenced by Lsm2p and Lsm4p, its presumed neighbours in the Lsm2-8p complex. Furthermore, overexpression and depletion experiments imply that Lsm1p and Lsm8p act competitively with respect to the localization of the two complexes, suggesting a potential mechanism for co-regulation of nuclear and cytoplasmic RNA processing. A shift of Lsm proteins from the nucleus to the cytoplasm under stress conditions indicates that this competition is biologically significant.
Agonists of kainate receptors (KARs) cause both the opening of the associated ion channels and the activation of signalling pathways driven by G-proteins and PKC. Here we report the existence of an unknown mechanism of KAR autoregulation, involving the interplay of this two signalling mechanisms. Repetitive activation of native KARs evoked the rundown of the ionotropic responses in a manner that was dependent on the activation of PKC. Experiments on recombinant GluR5 expressed in neuroblastoma cells indicated that KARs trigger the activation of PKC and induce the internalization of membrane receptors. This phenomenon depends on the PKC-mediated phosphorylation of serines 879 and 885 of the GluR5-2b subunits, since mutation of these two residues abolished internalization. These results reveal that the non-canonical signalling of KARs is associated with a sensitive mechanism that detects afferent activity. Such a mechanism represents an active way to limit overactivation of the KAR system, by regulating the number of KARs in the cell membrane.
The Kaposi's sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen (LANA) protein is functionally pleiotropic. LANA contributes to KSHV-associated pathogenesis, in part, by increasing entry of cells into S phase through a process that is driven by LANA interaction with the serine-threonine kinase glycogen synthase kinase 3 (GSK-3) and stabilization of beta-catenin. We now show that LANA affects the activity of another protein involved in cell cycle regulation, c-Myc. Sequencing of c-Myc coding sequences revealed that c-Myc in KSHV-positive primary effusion lymphoma (PEL) cell lines is wild type in the N-terminal region that regulates c-Myc protein stability. Despite this, c-Myc in PEL cells is stabilized. In LANA-expressing cells, inactivation of nuclear GSK-3 reduced phosphorylation of c-Myc at Thr58 and contributed to c-Myc stabilization by decreasing c-Myc ubiquitination. Phosphorylation of c-Myc on Ser62 also affects c-Myc stability and function. We now show that LANA increases the level of phosphorylated extracellular signal-regulated kinase 1 (ERK1) and increases ERK phosphorylation of c-Myc on Ser62. LANA also interacted with c-Myc, and c-Myc amino acids 147 to 220 were required for this interaction. LANA (L1006P) retained the ability to bind to c-Myc and activate ERK1, indicating that these events did not require LANA interaction with GSK-3. Thus, LANA stabilizes c-Myc; prevents the phosphorylation of c-Myc at Thr58, an event that promotes Myc-induced apoptosis; and independently stimulates phosphorylation of c-Myc at Ser62, an event that transcriptionally activates c-Myc. LANA-mediated manipulation of c-Myc function is likely to contribute to KSHV-associated tumorigenesis through the induction of c-Myc regulated cellular genes, as well as by the stimulation of cell cycle progression.
The emergence of chloroquine-resistant Plasmodium falciparum malaria imperils the lives of millions of people in Africa, Southeast Asia and South America. Chloroquine resistance is associated with mutations in the P. falciparum chloroquine resistance transporter (PfCRT). We expressed chloroquine-sensitive (HB3) and resistant (Dd2) pfcrt alleles in HEK293 human embryonic kidney cells. PfCRT localized to the lysosomal limiting membrane and was not detected in the plasma membrane. We observed significant acidification of lysosomes containing PfCRT HB3 and Dd2, with Dd2 acidifying significantly more than HB3. A mutant HB3 allele expressing the K76T mutation (earlier found to be key for chloroquine resistance) acidified to the same extent as Dd2, whereas the acidification by a Dd2 allele expressing the T76K "back mutation" was significantly less than Dd2. Thus, the amino acid at position 76 is both an important determinant of chloroquine resistance in parasites and of lysosomal acidification following heterologous expression. PfCRT may be capable of modulating the pH of the parasite digestive vacuole, and thus chloroquine availability. Chloroquine accumulation and glycyl-phenylalanine-2-naphthylamide-induced release of lysosomal Ca(2+) stores were unaffected by PfCRT expression. Cytoplasmic domain mutations did not alter PfCRT sorting to the lysosomal membrane. This heterologous expression system will be useful to characterize PfCRT protein structure and function, and elucidate its molecular role in chloroquine resistance.
Upon activation by far-red light, phytochrome A signals are transduced through several pathways to promote photomorphogenesis. The COP1 E3 ligase represses photomorphogenesis in part by targeting transcription activators such as LAF1 and HY5 for destruction. Another positive regulator of photomorphogenesis is HFR1, a basic helix-loop-helix (bHLH) transcription factor. Here, we show that HFR1 colocalizes with COP1 in nuclear bodies, and that the HFR1 N-terminal region (amino acids 1-131) interacts with the COP1 WD40 domain. HFR1(DeltaN), an HFR1 mutant lacking the two N-terminal, COP1-interacting motifs, still localizes in nuclear bodies and retains weak affinity for COP1. Both HFR1 and HFR1(DeltaN) can be ubiquitinated by COP1, although with different efficiencies. Expression of 35S-HFR1(DeltaN) in wild-type plants confers greater hypersensitivity to FR than 35S-HFR1 expression, and only seedlings expressing 35S-HFR1(DeltaN) display constitutive photomorphogenesis. These phenotypic differences can be attributed to the instability of HFR1 compared with HFR1(DeltaN). In transgenic plants, HFR1 levels are significantly elevated upon induced expression of a dominant-negative COP1 mutant that interferes with endogenous COP1 E3 activity. Moreover, induced expression of wild-type COP1 in transgenic plants accelerates post-translational degradation of HFR1 under FR light. Taken together, our results show that HFR1 is ubiquitinated by COP1 E3 ligase and marked for post-translational degradation during photomorphogenesis.
In plants, the cell wall is a major determinant of cell morphogenesis. Cell enlargement depends on the tightly regulated expansion of the wall, which surrounds each cell. However, the qualitative and quantitative mechanisms controlling cell wall enlargement are still poorly understood. Here, we report the molecular and functional characterization of LRX1, a new Arabidopsis gene that encodes a chimeric leucine-rich repeat/extensin protein. LRX1 is expressed in root hair cells and the protein is specifically localized in the wall of the hair proper, where it becomes insolubilized during development. lrx1-null mutants, isolated by a reverse-genetic approach, develop root hairs that frequently abort, swell, or branch. Complementation and overexpression experiments using modified LRX1 proteins indicate that the interaction with the cell wall is important for LRX1 function. These results suggest that LRX1 is an extracellular component of a mechanism regulating root hair morphogenesis and elongation by controlling either polarized growth or cell wall formation and assembly.
The establishment of precise topographic maps during neural development is facilitated by the presorting of axons in the pathway before they reach their targets. In the vertebrate visual system, such topography is seen clearly in the optic tract (OT) and in the optic radiations. However, the molecular mechanisms involved in pretarget axon sorting are poorly understood. Here, we show in zebrafish that the RNA-binding protein Hermes, which is expressed exclusively in retinal ganglion cells (RGCs), is involved in this process. Using a RiboTag approach, we show that Hermes acts as a negative translational regulator of specific mRNAs in RGCs. One of these targets is the guidance cue receptor Neuropilin 1 (Nrp1), which is sensitive to the repellent cue Semaphorin 3A (Sema3A). Hermes knock-down leads to topographic missorting in the OT through the upregulation of Nrp1. Restoring Nrp1 to appropriate levels in Hermes-depleted embryos rescues this effect and corrects the axon-sorting defect in the OT. Our data indicate that axon sorting relies on Hermes-regulated translation of Nrp1.
SIGNIFICANCE STATEMENT:
An important mechanism governing the formation of the mature neural map is pretarget axon sorting within the sensory tract; however, the molecular mechanisms involved in this process remain largely unknown. The work presented here reveals a novel function for the RNA-binding protein Hermes in regulating the topographic sorting of retinal ganglion cell (RGC) axons in the optic tract and tectum. We find that Hermes negatively controls the translation of the guidance cue receptor Neuropilin-1 in RGCs, with Hermes knock-down resulting in aberrant growth cone cue sensitivity and axonal topographic misprojections. We characterize a novel RNA-based mechanism by which axons restrict their translatome developmentally to achieve proper targeting.
Copyright © 2016 Hörnberg, Cioni et al.
AIMS:
Recent attempts to study MYC distribution in human samples have been confounded by a lack of agreement in immunohistochemical staining between antibodies targeting the N-terminus and those targeting the C-terminus of the MYC protein. The aim of this study was to use a novel in-situ hybridization (ISH) approach to detect MYC mRNA in clinically relevant samples, and thereby determine the reliability of MYC-targeting antibodies.
METHODS AND RESULTS:
We performed immunohistochemistry on human formalin-fixed paraffin embedded normal colon (n = 15), hyperplastic polyp (n = 4) and neoplastic colon samples (n = 55), using the N-terminally directed antibody Y69, and the C-terminally directed antibody 9E10. The MYC protein distributions were then compared with the location of MYC mRNA, determined by ISH. We found that the localization of MYC mRNA correlated well with the protein distribution determined with the N-terminally directed antibody Y69, and was also associated with expression of the proliferation marker Ki67. The protein distribution determined with the C-terminally directed antibody 9E10 was not always associated with MYC mRNA, Y69, or Ki67, and indeed often showed a reciprocal pattern of expression, with staining being strongest in non-proliferating cells.
CONCLUSIONS:
The observed discrepancy between the staining patterns suggests that the significance of 9E10 in immunohistochemical staining is currently uncertain, and therefore should be interpreted with caution.
© 2016 The Authors. Histopathology published by John Wiley & Sons Ltd.
BACKGROUND:
Heterochromatin is essential for chromosome segregation, gene silencing and genome integrity. The fission yeast Schizosaccharomyces pombe contains heterochromatin at centromeres, subtelomeres, and mating type genes, as well as at small islands of meiotic genes dispersed across the genome. This heterochromatin is generated by partially redundant mechanisms, including the production of small interfering RNAs (siRNAs) that are incorporated into the RITS protein complex (RNAi-Induced Transcriptional Silencing). The assembly of heterochromatin islands requires the function of the RNA-binding protein Mmi1, which recruits RITS to its mRNA targets and to heterochromatin islands. In addition, Mmi1 directs its targets to an exosome-dependent RNA elimination pathway.
RESULTS:
Ccr4-Not is a conserved multiprotein complex that regulates gene expression at multiple levels, including RNA degradation and translation. We show here that Ccr4-Not is recruited by Mmi1 to its RNA targets. Surprisingly, Ccr4 and Caf1 (the mRNA deadenylase catalytic subunits of the Ccr4-Not complex) are not necessary for the degradation or translation of Mmi1 RNA targets, but are essential for heterochromatin integrity at Mmi1-dependent islands and, independently of Mmi1, at subtelomeric regions. Both roles require the deadenylase activity of Ccr4 and the Mot2/Not4 protein, a ubiquitin ligase that is also part of the complex. Genetic evidence shows that Ccr4-mediated silencing is essential for normal cell growth, indicating that this novel regulation is physiologically relevant. Moreover, Ccr4 interacts with components of the RITS complex in a Mmi1-independent manner.
CONCLUSIONS:
Taken together, our results demonstrate that the Ccr4-Not complex is required for heterochromatin integrity in both Mmi1-dependent and Mmi1-independent pathways.
BACKGROUND:
Brewers' rice, is locally known as temukut, is a mixture of broken rice, rice bran, and rice germ. The current study is an extension of our previous work, which demonstrated that water extract of brewers' rice (WBR) induced apoptosis in human colorectal cancer (HT-29) cells. We also identified that brewers' rice was effective in reducing the tumor incidence and multiplicity in azoxymethane (AOM)-injected colon cancer rats. Our present study was designed to identify whether WBR confers an inhibitory effect via the regulation of upstream components in the Wnt signaling pathway in HT-29 cells. To further determine whether the in vitro mechanisms of action observed in the HT-29 cells inhibit the downstream signaling target of the Wnt/β-catenin pathway, we evaluated the mechanistic action of brewers' rice in regulating the expressions and key protein markers during colon carcinogenesis in male Sprague-Dawley rats.
METHODS:
The mRNA levels of several upstream-related genes in the Wnt signaling pathway in HT-29 cells treated with WBR were determined by quantitative real-time PCR analyses. Caspase-3 and -8 were evaluated using a colorimetric assay. Male Sprague-Dawley rats were administered two intraperitoneal injections of AOM in saline (15 mg/kg body weight) over a two-week period and received with 10, 20, and 40% (w/w) brewers' rice. The expressions and protein levels of cyclin D1 and c-myc were evaluated by immunohistochemical staining and western blotting, respectively.
RESULTS:
The overall analyses revealed that the treatment of HT-29 cells with WBR inhibited Wnt signaling activity through upregulation of the casein kinase 1 (CK1) and adenomatous polyposis coli (APC) mRNA levels. We discovered that the treatment of HT-29 cells with WBR resulted in the induction of apoptosis by the significant activation of caspase-3 and -8 activities compared with the control (P < 0.05). In vivo analyses indicated that brewers' rice diminished the β-catenin, cyclin D1, and c-myc protein levels.
CONCLUSIONS:
We provide evidence that brewers' rice can induce apoptosis and inhibit the proliferation of HT-29 cells through regulation of caspase-dependent pathways and inhibit the Wnt/β-catenin downstream signaling pathway in vivo. We suggest that brewers' rice may be a useful dietary agent for colorectal cancer.
BACKGROUND:
An increasing number of evidence suggests that pancreatic cancer contains cancer stem cells (CSCs), which may be relevant to the resistance of chemotherapy. Latexin (Lxn) is a negative regulator of stem cell proliferation and we investigate the effects of Lxn on CD133+ pancreatic cancer stem-like cells.
METHODS:
CD133+ miapaca-2 cells, a human pancreatic carcinoma cell line, were isolated and sorted by magnetic activated cell sorting and flow cytometry. The capacity for self-renewal, proliferation, and tumorigenicity of CD133+ miapaca-2 cells was determined by the floating spheres test and tumor xenograft assays. Protein and mRNA expression of Lxn in CD133+ and CD133- miapaca-2 cells were detected by Western blotting and qRT-PCR, respectively. After CD133+ miapaca-2 cells were treated with Lxn in serum-free medium (SFM), cell proliferation was assayed with a Cell Counting Kit 8 (CCK-8) and apoptosis was analyzed by flow cytometry. The protein and mRNA expression levels of Bcl-2, bax, and c-myc were also analyzed.
RESULTS:
We successfully isolated CD133+ miapaca-2 cells that exhibited the capacity for self-renewal in SFM, a proliferation potential in DMEM supplemented with FBS, and high tumorigenicity in nude mice. Lxn protein and mRNA expression levels in CD133+ miapaca-2 cells were significantly lower than those in CD133- cells. Lxn-treated CD133+ miapaca-2 cells exhibited increased apoptosis and low proliferation activity, down-regulation of Bcl-2 and c-myc expression, and up-regulation of Bax expression in a dose-dependent manner.
CONCLUSIONS:
Lxn induces apoptosis and inhibits the proliferation of CD133+ miapaca-2 cells. These changes are associated with down-regulation of Bcl-2 and c-myc and up-regulation of Bax.
BACKGROUND:
IQ-domain GTPase-activating protein 1 (IQGAP1) binds to Dishevelled (Dvl) and functions as a modulator of Dvl nuclear localization in Xenopus embryos. However, the relationship between IQGAP1 and Dvl in tumor tissues is unclear.
MATERIALS AND METHODS:
We used immunohistochemistry to assess the expressions of IQGAP1 and Dvl in a cohort of 111 non-small cell lung cancer (NSCLC) patients. Association of their localization expressions with clinicopathological factors was also analyzed.
RESULTS:
The positive rate of IQGAP1 in primary tumors was 48.6% (54/111) for its cytoplamic expression, 9.0% (10/111) for nuclear expression and 31.5% (35/111) for membranous expression; the positive rate of Dvl was 65.8% (73/111) for cytoplamic expression, 9.9% (11/111) for nuclear expression and 10.8% (12/111) for membranous expression. Coexpression rate of IQGAP1 and Dvl was 77.8% (42/54) in the cytoplasm, 80.0% (8/10) in the nucleus and 8.6% (3/35) in the membrane. Coexpression of IQGAP1 and Dvl in the cytoplasm and nucleus were significantly correlated (P<0.05), but not in the membrane (P>0.05). The positive expression rates of cyclin D1 and c-myc were significantly higher in the group of IQGAP1 and Dvl coexpression in the nucleus than that in the cytoplasm. Coexpression rate of IQGAP1 and Dvl in the cytoplasm and nucleus was significantly higher in lymph nodal metastases (63.3%, 19/30) than in primary growths (38.3%, 31/81), correlating with poor prognosis. Five-year survival time after resection in the group with their coexpression in the cytoplasm and nucleus was significantly lower than that with no coexpression (44.705±3.355 vs 58.403±2.543 months, p<0.05).
CONCLUSIONS:
Coexpression of IQGAP1 and Dvl in the cytoplasm and nucleus was correlated with the lymph nodal metastase and poor prognosis of NSCLC, and coexpression in nucleus might play a critical role in the activation of canonical Wnt pathway.
BACKGROUND:
MicroRNA (miRNA, miR)-18a is a member of the miR-17-92 cluster, an important locus that is markedly overexpressed in several cancers and associated with cancer development and progression. However, the mechanism of action of the miR-17-92 cluster and its individual miRNAs are largely unknown.
METHODS AND RESULTS:
In this study, we investigated the expression of the miR-17-92 cluster by in situ hybridisation (ISH) assay and copy-number analysis in gastric tissue microarray (TMA) specimens. We determined that miR-18a was present at higher levels than the other five miRNAs in the cluster. In addition, we identified Protein Inhibitor of Activated Signal Transducer and Activator of Transcription 3 (PIAS3) as a direct target of miR-18a in gastric cancer. miR-18a level was positively correlated with levels of Survivin, Bcl-xL, and c-Myc, which are downstream transcriptional targets of Signal Transducer and Activator of Transcription 3 (STAT3). STAT3-induced transcription can be negatively regulated by PIAS3; consistent with this, PIAS3 level was negatively correlated with levels of Survivin, Bcl-xL, and c-Myc.
CONCLUSION:
Our findings indicate that miR-18a acts as an oncogene and plays a role in gastric adenocarcinogenesis, at least in part by negatively regulating PIAS3 and thereby modulating expression of STAT3 target genes.
INTRODUCTION:
Overexpression of the Polycomb repressive complex 2 (PRC2) subunit Enhancer of Zeste 2 (EZH2) occurs in several malignancies, including prostate cancer, breast cancer, medulloblastoma, and glioblastoma multiforme. Recent evidence suggests that EZH2 may also have a role in rhabdoid tumors. Atypical teratoid/rhabdoid tumor (ATRT) is a rare, high-grade embryonal brain tumor that occurs most commonly in young children and carries a very poor prognosis. ATRTs are characterized by absence of the chromatin remodeling protein SMARCB1. Given the role of EZH2 in regulating epigenetic changes, we investigated the role of EZH2 in ATRT.
METHODS:
Microarray analysis was used to evaluate expression of EZH2 in ATRT tumor samples. We used shRNA and a chemical inhibitor of EZH2 to examine the impact of EZH2 inhibition on cell growth, proliferation, and tumor cell self-renewal.
RESULTS:
Here, we show that targeted disruption of EZH2 by RNAi or pharmacologic inhibition strongly impairs ATRT cell growth, suppresses tumor cell self-renewal, induces apoptosis, and potently sensitizes these cells to radiation. Using functional analysis of transcription factor activity, we found the cyclin D1-E2F axis to be repressed after EZH2 depletion in ATRT cells.
CONCLUSIONS:
Our observations provide evidence that EZH2 disruption alters cell cycle progression and may be an important new therapeutic target, particularly in combination with radiation, in ATRT.
BACKGROUND:
Cancer stem cells (CSCs) play an important role in cancer initiation, relapse and metastasis. To date, no specific medicine has been found to target CSCs as they are resistant to most conventional therapies and proliferate indefinitely. Compound Kushen Injection (CKI) has been widely used for cancer patients with remarkable therapeutic effects in Chinese clinical settings for many years. This study focused on whether CKI could inhibit MCF-7 SP cells in vitro and in vivo.
METHODS:
The analysis of CKI on SP population and the main genes of Wnt signaling pathway were studied first. Then we studied the tumorigenicity of SP cells and the effects of CKI on SP cells in vivo. The mice inoculated with 10,000 SP cells were randomly divided into three groups (6 in each group) and treated with CKI, cisplatin and saline (as a control) respectively for 7 weeks. The tumor formation rates of each group were compared. The main genes and proteins of the Wnt signaling pathway were analyzed by RT-PCR and western blot.
RESULTS:
CKI suppressed the size of SP population (approximately 90%), and down-regulated the main genes of Wnt signaling pathway. We also determined that MCF-7 SP cells were more tumorigenic than non-SP and unsorted cells. The Wnt signaling pathway was up-regulated in tumors derived from SP cells compared with that in tumors from non-SP cells. The tumor formation rate of the CKI Group was 33% (2/6, P < 0.05), and that of Cisplatin Group was 50%(3/6, P < 0.05), whereas that of the Control Group was 100% (6/6).The RT-PCR and western blot results indicated that CKI suppressed tumor growth by down-regulating the Wnt/β-catenin pathway, while cisplatin activated the Wnt/β-catenin pathway and might spare SP cells.
CONCLUSIONS:
It suggested that CKI may serve as a novel drug targeting cancer stem-like cells, though further studies are recommended.
BACKGROUND:
Oligomerization and aggregation of alpha-synuclein molecules play a major role in neuronal dysfunction and loss in Parkinson's disease [1]. However, alpha-synuclein oligomerization and aggregation have mostly been detected indirectly in cells using detergent extraction methods [2], [3], [4]. A number of in vitro studies showed that dopamine can modulate the aggregation of alpha-synuclein by inhibiting the formation of or by disaggregating amyloid fibrils [5], [6], [7].
METHODOLOGY/PRINCIPAL FINDINGS:
Here, we show that alpha-synuclein adopts a variety of conformations in primary neuronal cultures using fluorescence lifetime imaging microscopy (FLIM). Importantly, we found that dopamine, but not dopamine agonists, induced conformational changes in alpha-synuclein which could be prevented by blocking dopamine transport into the cell. Dopamine also induced conformational changes in alpha-synuclein expressed in neuronal cell lines, and these changes were also associated with alterations in oligomeric/aggregated species.
CONCLUSION/SIGNIFICANCE:
Our results show, for the first time, a direct effect of dopamine on the conformation of alpha-synuclein in neurons, which may help explain the increased vulnerability of dopaminergic neurons in Parkinson's disease.
BACKGROUND:
All human pathogenic Yersinia species share a virulence-associated type III secretion system that translocates Yersinia effector proteins into host cells to counteract infection-induced signaling responses and prevent phagocytosis. Dictyostelium discoideum has been recently used to study the effects of bacterial virulence factors produced by internalized pathogens. In this study we explored the potential of Dictyostelium as model organism for analyzing the effects of ectopically expressed Yersinia outer proteins (Yops).
RESULTS:
The Yersinia pseudotuberculosis virulence factors YopE, YopH, YopM and YopJ were expressed de novo within Dictyostelium and their effects on growth in axenic medium and on bacterial lawns were analyzed. No severe effect was observed for YopH, YopJ and YopM, but expression of YopE, which is a GTPase activating protein for Rho GTPases, was found to be highly detrimental. GFP-tagged YopE expressing cells had less conspicuous cortical actin accumulation and decreased amounts of F-actin. The actin polymerization response upon cAMP stimulation was impaired, although chemotaxis was unaffected. YopE also caused reduced uptake of yeast particles. These alterations are probably due to impaired Rac1 activation. We also found that YopE predominantly associates with intracellular membranes including the Golgi apparatus and inhibits the function of moderately overexpressed RacH.
CONCLUSION:
The phenotype elicited by YopE in Dictyostelium can be explained, at least in part, by inactivation of one or more Rho family GTPases. It further demonstrates that the social amoeba Dictyostelium discoideum can be used as an efficient and easy-to-handle model organism in order to analyze the function of a translocated GAP protein of a human pathogen.
BACKGROUND:
The primary cilium is a sensory organelle generated from the centrosome in quiescent cells and found at the surface of most cell types, from where it controls important physiological processes. Specific sets of membrane proteins involved in sensing the extracellular milieu are concentrated within cilia, including G protein coupled receptors (GPCRs). Most GPCRs are regulated by beta-arrestins, betaarr1 and betaarr2, which control both their signalling and endocytosis, suggesting that betaarrs may also function at primary cilium.
METHODOLOGY/PRINCIPAL FINDINGS:
In cycling cells, betaarr2 was observed at the centrosome, at the proximal region of the centrioles, in a microtubule independent manner. However, betaarr2 did not appear to be involved in classical centrosome-associated functions. In quiescent cells, both in vitro and in vivo, betaarr2 was found at the basal body and axoneme of primary cilia. Interestingly, betaarr2 was found to interact and colocalize with 14-3-3 proteins and Kif3A, two proteins known to be involved in ciliogenesis and intraciliary transport. In addition, as suggested for other centrosome or cilia-associated proteins, betaarrs appear to control cell cycle progression. Indeed, cells lacking betaarr2 were unable to properly respond to serum starvation and formed less primary cilia in these conditions.
CONCLUSIONS/SIGNIFICANCE:
Our results show that betaarr2 is localized to the centrosome in cycling cells and to the primary cilium in quiescent cells, a feature shared with other proteins known to be involved in ciliogenesis or primary cilium function. Within cilia, betaarr2 may participate in the signaling of cilia-associated GPCRs and, therefore, in the sensory functions of this cell "antenna".
Neural cell fate acquisition is mediated by transcription factors expressed in nascent neuroectoderm, including Geminin and members of the Zic transcription factor family. However, regulatory networks through which this occurs are not well defined. Here, we identified Geminin-associated chromatin locations in embryonic stem cells and Geminin- and Zic1-associated locations during neural fate acquisition at a genome-wide level. We determined how Geminin deficiency affected histone acetylation at gene promoters during this process. We integrated these data to demonstrate that Geminin associates with and promotes histone acetylation at neurodevelopmental genes, while Geminin and Zic1 bind a shared gene subset. Geminin- and Zic1-associated genes exhibit embryonic nervous system-enriched expression and encode other regulators of neural development. Both Geminin and Zic1-associated peaks are enriched for Zic1 consensus binding motifs, while Zic1-bound peaks are also enriched for Sox3 motifs, suggesting co-regulatory potential. Accordingly, we found that Geminin and Zic1 could cooperatively activate the expression of several shared targets encoding transcription factors that control neurogenesis, neural plate patterning, and neuronal differentiation. We used these data to construct gene regulatory networks underlying neural fate acquisition. Establishment of this molecular program in nascent neuroectoderm directly links early neural cell fate acquisition with regulatory control of later neurodevelopment.
Deacetylation of α-tubulin at lysine 40 is catalyzed by two enzymes, the NAD-dependent deacetylase SIRT2 and the NAD-independent deacetylase HDAC6, in apparently redundant reactions. In the present study, we tested whether these two enzymes might have distinguishable preferences for the deacetylation of different microtubule structures. Using various agents, we induced tubulin hyperacetylation and analyzed the ensuing formation of distinct microtubule structures. HDAC6 inhibition led to general hyperacetylation of the microtubule network throughout the cell, whereas hyperacetylation induced by SIRT2 inactivation was limited to perinuclear microtubules. Hyperacetylation of these perinuclear microtubules was undiminished following HDAC6 overexpression, whereas reactivation of SIRT2 restored the basal acetylation level and a normal microtubule network. By contrast, SIRT2 and HDAC6 acted similarly on the morphologically different, hyperacetylated microtubule structures induced by taxol, MAP2c overexpression or hyperosmotic stress. These results indicate overlapping and distinct functions of HDAC6 and SIRT2. We propose that the differential activity of the two deacetylases, which target the same acetylated lysine residue, might be related to the recognition of specific structural contexts.
Regulation of STAT3 activation is critical for normal and malignant hematopoietic cell proliferation. Here, we have reported that the endogenous transmembrane protein upstream-of-mTORC2 (UT2) negatively regulates activation of STAT3. Specifically, we determined that UT2 interacts directly with GP130 and inhibits phosphorylation of STAT3 on tyrosine 705 (STAT3Y705). This reduces cytokine signaling including IL6 that is implicated in multiple myeloma and other hematopoietic malignancies. Modulation of UT2 resulted in inverse effects on animal survival in myeloma models. Samples from multiple myeloma patients also revealed a decreased copy number of UT2 and decreased expression of UT2 in genomic and transcriptomic analyses, respectively. Together, these studies identify a transmembrane protein that functions to negatively regulate cytokine signaling through GP130 and pSTAT3Y705 and is molecularly and mechanistically distinct from the suppressors of cytokine signaling (SOCS) family of genes. Moreover, this work provides evidence that perturbations of this activation-dampening molecule participate in hematologic malignancies and may serve as a key determinant of multiple myeloma pathophysiology. UT2 is a negative regulator shared across STAT3 and mTORC2 signaling cascades, functioning as a tumor suppressor in hematologic malignancies driven by those pathways.
Analysis of embryonic fibroblasts from GFP reporter mice indicates that the fibroblast cell type harbors a large collection of developmentally and phenotypically heterogeneous subtypes. Some of these cells exhibit multipotency, whereas others do not. Multiparameter flow cytometry analysis shows that a large number of distinct populations of fibroblast-like cells can be found in cultures initiated from different embryonic organs, and cells sorted according to their surface phenotype typically retain their characteristics on continued propagation in culture. Similarly, surface phenotypes of individual cloned fibroblast-like cells exhibit significant variation. The fibroblast cell class appears to contain a very large number of denumerable subtypes.
The Popeye domain-containing 1 (POPDC1) gene encodes a plasma membrane-localized cAMP-binding protein that is abundantly expressed in striated muscle. In animal models, POPDC1 is an essential regulator of structure and function of cardiac and skeletal muscle; however, POPDC1 mutations have not been associated with human cardiac and muscular diseases. Here, we have described a homozygous missense variant (c.602C>T, p.S201F) in POPDC1, identified by whole-exome sequencing, in a family of 4 with cardiac arrhythmia and limb-girdle muscular dystrophy (LGMD). This allele was absent in known databases and segregated with the pathological phenotype in this family. We did not find the allele in a further screen of 104 patients with a similar phenotype, suggesting this mutation to be family specific. Compared with WT protein, POPDC1(S201F) displayed a 50% reduction in cAMP affinity, and in skeletal muscle from patients, both POPDC1(S201F) and WT POPDC2 displayed impaired membrane trafficking. Forced expression of POPDC1(S201F) in a murine cardiac muscle cell line (HL-1) increased hyperpolarization and upstroke velocity of the action potential. In zebrafish, expression of the homologous mutation (popdc1(S191F)) caused heart and skeletal muscle phenotypes that resembled those observed in patients. Our study therefore identifies POPDC1 as a disease gene causing a very rare autosomal recessive cardiac arrhythmia and LGMD, expanding the genetic causes of this heterogeneous group of inherited rare diseases.
In human cancers, β-catenin is accumulated in the nucleus and activates mRNA transcription of many oncogenic genes, such as cyclin D1 and c-myc. However, the mechanism of β-catenin-mediated transcriptional activation remains largely unknown. In the present study, we identified leupaxin, an adaptor protein sharing homology with the focal adhesion protein, as a novel coactivator for β-catenin in human hepatocellular carcinoma (HCC). We show that leupaxin could interact with β-catenin and enhance its transcriptional activity through recruitment of coactivator complex, including steroid receptor coactivator 1 (SRC-1) and P300. As a result, leupaxin regulates HCC cell proliferation and cell-cycle progression in the presence of intact Wnt/β-catenin signaling. Furthermore, leupaxin is overexpressed in HCC tissues and correlated with mRNA levels of cyclin D1 and c-myc. Therefore, this is the first demonstration of a role for the leupaxin in the regulation of HCC progression, at least in part, by enhancing β-catenin transcription activity.
Gremlin is a member of the CAN (cerberus and DAN) family of secreted BMP (bone morphogenetic protein) antagonists and also an agonist of VEGF (vascular endothelial growth factor) receptor-2. It is critical in limb skeleton and kidney development and is re-expressed during tissue fibrosis. Gremlin binds strongly to heparin and heparan sulfate and, in the present study, we sought to investigate its heparin-binding site. In order to explore a putative non-contiguous binding site predicted by computational molecular modelling, we substituted a total of 11 key arginines and lysines located in three basic residue sequence clusters with homologous sequences from cerberus and DAN (differential screening selected gene abberative in neuroblastoma), CAN proteins which lack basic residues in these positions. A panel of six Myc-tagged gremlin mutants, MGR-1-MGR-6 (MGR, mutant gremlin), each containing different combinations of targeted substitutions, all showed markedly reduced affinity for heparin as demonstrated by their NaCl elution on heparin affinity chromatography, thus verifying our predictions. Both MGR-5 and MGR-6 retained BMP-4-binding activity comparable to that of wild-type gremlin. Low-molecular-mass heparin neither promoted nor inhibited BMP-4 binding. Finally, glutaraldehyde cross-linking demonstrated that gremlin forms non-covalent dimers, similar behaviour to that of DAN and also PRDC (protein related to cerberus and DAN), another CAN protein. The resulting dimer would possess two heparin-binding sites, each running along an exposed surface on the second β-strand finger loop of one of the monomers.
The non-receptor-type tyrosine kinase c-Abl functions as a cytoplasmic signal transducer upon activation of cell-surface receptors. c-Abl is also involved in DDR (DNA-damage response), which is initiated in the nucleus, whereas its molecular functions in DDR are not fully understood. In the present study, we found that c-Abl phosphorylates JunB, a member of the AP-1 (activator protein 1) transcription factor family. Because JunB was suggested to be involved in DDR, we analysed the role of c-Abl-mediated phosphorylation of JunB in DDR. We first analysed phosphorylation sites of JunB and found that c-Abl majorly phosphorylates JunB at Tyr(173), Tyr(182) and Tyr(188). Because c-Abl promotes expression of the cyclin-dependent kinase inhibitor p21 upon stimulation with the DNA-damaging agent Adriamycin (doxorubicin), we analysed the involvement of JunB in Adriamycin-induced p21 expression. We found that JunB suppresses p21 induction through inhibition of its promoter activity. The phosphomimetic JunB, which was generated by glutamic acid substitutions at the phosphorylation sites, failed to repress p21 induction. Recruitment of JunB to the p21 promoter was promoted by Adriamycin stimulation and was further enhanced by co-treatment with the c-Abl inhibitor imatinib. The phosphomimetic glutamic acid substitutions in JunB or Adriamycin treatment impaired the JunB-c-Fos transcription factor complex formation. Taken together, these results suggest that, although JunB represses p21 promoter activity, c-Abl phosphorylates JunB and conversely inhibits its suppressive role on p21 promoter activity upon Adriamycin stimulation. Therefore JunB is likely to be a key target of c-Abl in expression of p21 in Adriamycin-induced DDR.
Chemotherapy continues to be the standard treatment for advanced or metastasized cancer. However, commonly used chemotherapeutic agents may induce damage in healthy cells and tissues. Thus, in recent years, there has been an increased focus on the development of new, efficient anticancer drugs exhibiting low toxicity and that are not affected by mechanisms of chemoresistance. In the present work, we tested synthetic and naturally obtained human salivary peptides against breast, prostate, colon, osteosarcoma and bladder cancer cell lines (T47-D, PC-3, HT-29, MG63, T-24, respectively). Results have showed that there is a reduced cell population increase that is peptide-, cell- and possibly pathway-specific, with the most potent effect observed in observed in T-47D breast cancer cells. Protein expression and microscopy results further indicate that, in this cell line, the peptide with the sequence GPPPQGGRPQG (GG peptide) interferes with the ability of cell adhesion proteins to stabilize adherens junctions, such as E-cadherin, leading to apoptosis. These promising results encourage future works aimed at disclosing the vast potential of salivary peptides as new therapeutic agents.
The study of cell-surface receptor dynamics is critical for understanding how cells sense and respond to changing environments. Therefore, elucidating the mechanisms by which signals are perceived and communicated into the cell is necessary to understand immunity, development, and stress. Challenges in testing interactions of membrane-bound proteins include their dynamic nature, their abundance, and the complex dual environment (lipid/soluble) in which they reside. Co-Immunoprecipitation (Co-IP) of tagged membrane proteins is a widely used approach to test protein-protein interaction in vivo. In this protocol we present a method to perform Co-IP using enriched membrane proteins in isolated microsomal fractions. The different variations of this protocol are highlighted, including recommendations and troubleshooting guides in order to optimize its application. This Co-IP protocol has been developed to test the interaction of receptor-like kinases, their interacting partners, and peptide ligands in stable Arabidopsis thaliana lines, but can be modified to test interactions in transiently expressed proteins in tobacco, and potentially in other plant models, or scaled for large-scale protein-protein interactions at the membrane.
In response to chromosomal double-strand breaks (DSBs), eukaryotic cells activate the DNA damage checkpoint, which is orchestrated by the PI3 kinase-like protein kinases ATR and ATM (Mec1 and Tel1 in budding yeast). Following DSB formation, Mec1 and Tel1 phosphorylate histone H2A on serine 129 (known as γ-H2AX). We used caffeine to inhibit the checkpoint kinases after DSB induction. We show that prolonged phosphorylation of H2A-S129 does not require continuous Mec1 and Tel1 activity. Unexpectedly, caffeine treatment impaired homologous recombination by inhibiting 5' to 3' end resection, independent of Mec1 and Tel1 inhibition. Caffeine treatment led to the rapid loss, by proteasomal degradation, of both Sae2, a nuclease that plays a role in early steps of resection, and Dna2, a nuclease that facilitates one of two extensive resection pathways. Sae2's instability is evident in the absence of DNA damage. A similar loss is seen when protein synthesis is inhibited by cycloheximide. Caffeine treatment had similar effects on irradiated HeLa cells, blocking the formation of RPA and Rad51 foci that depend on 5' to 3' resection of broken chromosome ends. Our findings provide insight toward the use of caffeine as a DNA damage-sensitizing agent in cancer cells.
During immune reactions, functionally distinct B-cell subsets are generated by stochastic processes, including class-switch recombination (CSR) and plasma cell differentiation (PCD). In this study, we show a strong association between individual B-cell fates and mitochondrial functions. CSR occurs specifically in activated B cells with increased mitochondrial mass and membrane potential, which augment mitochondrial reactive oxygen species (mROS), whereas PCD occurs in cells with decreased mitochondrial mass and potential. These events are consequences of initial slight changes in mROS in mitochondria(high) B-cell populations. In CSR-committed cells, mROS attenuates haeme synthesis by inhibiting ferrous ion addition to protoporphyrin IX, thereby maintaining Bach2 function. Reduced mROS then promotes PCD by increasing haeme synthesis. In PCD-committed cells, Blimp1 reduces mitochondrial mass, thereby reducing mROS levels. Identifying mROS as a haeme synthesis regulator increases the understanding of mechanisms regulating haeme homeostasis and cell fate determination after B-cell activation.
Tau abnormalities play a central role in several neurodegenerative diseases, collectively known as tauopathies. In the present study, we examined whether mutant huntingtin (mHtt), which causes Huntington's disease (HD), modifies Tau phosphorylation and subcellular localization using cell and mouse HD models. Initially, we used novel bimolecular fluorescence complementation assays in live cells to evaluate Tau interactions with either wild type (25QHtt) or mutant huntingtin (103QHtt). While 25QHtt and Tau interacted at the level of the microtubule network, 103QHtt and Tau interacted and formed 'ring-like' inclusions localized in the vicinity of the microtubular organizing center (MTOC). Fluorescence recovery after photobleaching experiments also indicated that, whereas homomeric 103QHtt/103QHtt pairs rapidly re-entered into inclusions, heteromeric 103QHtt/Tau pairs remained excluded from the 'ring-like' inclusions. Interestingly, in vitro Tau relocalization was associated to Tau hyperphosphorylation. Consistent with this observation, we found strong Tau hyperphosphorylation in brain samples from two different mouse models of HD, R6/2 and 140CAG knock-in. This was associated with a significant reduction in the levels of Tau phosphatases (PP1, PP2A and PP2B), with no apparent involvement of major Tau kinases. Thus, the present study strongly suggests that expression of mHtt leads to Tau hyperphosphorylation, relocalization and sequestration through direct protein-protein interactions in inclusion-like compartments in the vicinity of the MTOC. Likewise, our data also suggest that Tau alterations may also contribute to HD pathogenesis.
New reliable and cost-effective antimalarial drug screening assays are urgently needed to identify drugs acting on different stages of the parasite Plasmodium falciparum, and particularly those responsible for human-to-mosquito transmission, that is, the P. falciparum gametocytes. Low Z' factors, narrow dynamic ranges, and/or extended assay times are commonly reported in current gametocyte assays measuring gametocyte-expressed fluorescent or luciferase reporters, endogenous ATP levels, activity of gametocyte enzymes, or redox-dependent dye fluorescence. We hereby report on a dual-luciferase gametocyte assay with immature and mature P. falciparum gametocyte stages expressing red and green-emitting luciferases from Pyrophorus plagiophthalamus under the control of the parasite sexual stage-specific pfs16 gene promoter. The assay was validated with reference antimalarial drugs and allowed to quantitatively and simultaneously measure stage-specific drug effects on parasites at different developmental stages. The optimized assay, requiring only 48 h incubation with drugs and using a cost-effective luminogenic substrate, significantly reduces assay cost and time in comparison to state-of-the-art analogous assays. The assay had a Z' factor of 0.71 ± 0.03, and it is suitable for implementation in 96- and 384-well microplate formats. Moreover, the use of a nonlysing D-luciferin substrate significantly improved the reliability of the assay and allowed one to perform, for the first time, P. falciparum bioluminescence imaging at single-cell level.
Correct chromosome segregation requires a unique chromatin environment at centromeres and in their vicinity. Here, we address how the deposition of canonical H2A and H2A.Z histone variants is controlled at pericentric heterochromatin (PHC). Whereas in euchromatin newly synthesized H2A and H2A.Z are deposited throughout the cell cycle, we reveal two discrete waves of deposition at PHC - during mid to late S phase in a replication-dependent manner for H2A and during G1 phase for H2A.Z. This G1 cell cycle restriction is lost when heterochromatin features are altered, leading to the accumulation of H2A.Z at the domain. Interestingly, compromising PHC integrity also impacts upon neighboring centric chromatin, increasing the amount of centromeric CENP-A without changing the timing of its deposition. We conclude that the higher-order chromatin structure at the pericentric domain influences dynamics at the nucleosomal level within centromeric chromatin. The two different modes of rearrangement of the PHC during the cell cycle provide distinct opportunities to replenish one or the other H2A variant, highlighting PHC integrity as a potential signal to regulate the deposition timing and stoichiometry of histone variants at the centromere.
Previous studies have established the link between aberrant microRNA (miRNA) expression and hypoxia in various neoplasms. However, how these hypoxia-related miRNAs modulate tumor progression is still unclear. Therefore, the patterns of miRNA in colorectal carcinoma cell lines in response to hypoxia or not were first screened and the hypoxia-induced repression of the miR-15-16 cluster was confirmed. Then, this repression was found to be associated with high tumor stage and poor prognosis in colorectal carcinoma and is shown to promote tumor angiogenesis and metastasis by the loss of restriction of its target gene, fibroblast growth factor-2 (FGF2). Moreover, the general and alterative promoters of the miR-15-16 host (deleted in lymphocytic leukemia 2, DLEU2) were mapped, and three c-Myc/Max binding sites in response to the hypoxia-induced repression of miR-15-16 were further identified. Finally, an enhanced stability of c-Myc/Max heterodimer promoted by increased hypoxia-inducible factor-2α (HIF-2α) was validated, and we also verified that the enhancement contributed to the hypoxia-induced repression of miR-15-16. In brief, the c-Myc-mediated transcriptional repression of miR-15-16 in hypoxia is induced by increased HIF-2α and promoted tumor angiogenesis and hematogenous metastasis by the further loss of post-transcriptional inhibition of FGF2. Our study provides a better understanding of the coping mechanisms in response to tumor hypoxia and may be helpful in developing an effective prognostic marker or treatment target against solid tumors.
Entry into mitosis is triggered by activation of Cdk1 and inactivation of its counteracting phosphatase PP2A/B55. Greatwall kinase inactivates PP2A/B55 via its substrates Ensa and ARPP19. Both Greatwall and Ensa/ARPP19 are regulated by phosphorylation, but the dynamic regulation of Greatwall activity and the phosphatases that control Greatwall kinase and its substrates are poorly understood. To address these questions we applied a combination of mathematical modelling and experiments using phospho-specific antibodies to monitor Greatwall, Ensa/ARPP19 and Cdk substrate phosphorylation during mitotic entry and exit. We demonstrate that PP2A/B55 is required for Gwl dephosphorylation at the essential Cdk site Thr194. Ensa/ARPP19 dephosphorylation is mediated by the RNA Polymerase II carboxy terminal domain phosphatase Fcp1. Surprisingly, inhibition or depletion of neither Fcp1 nor PP2A appears to block dephosphorylation of the bulk of mitotic Cdk1 substrates during mitotic exit. Taken together our results suggest a hierarchy of phosphatases coordinating Greatwall, Ensa/ARPP19 and Cdk substrate dephosphorylation during mitotic exit.
Autophagy is reported to be an important innate immune defense against the intracellular bacterial pathogen Group A Streptococcus (GAS). However, the GAS strains examined to date belong to serotypes infrequently associated with human disease. We find that the globally disseminated serotype M1T1 clone of GAS can evade autophagy and replicate efficiently in the cytosol of infected cells. Cytosolic M1T1 GAS (strain 5448), but not M6 GAS (strain JRS4), avoids ubiquitylation and recognition by the host autophagy marker LC3 and ubiquitin-LC3 adaptor proteins NDP52, p62, and NBR1. Expression of SpeB, a streptococcal cysteine protease, is critical for this process, as an isogenic M1T1 ΔspeB mutant is targeted to autophagy and attenuated for intracellular replication. SpeB degrades p62, NDP52, and NBR1 in vitro and within the host cell cytosol. These results uncover a proteolytic mechanism utilized by GAS to escape the host autophagy pathway that may underpin the success of the M1T1 clone.
In neurons, the timely and accurate expression of genes in response to synaptic activity relies on the interplay between epigenetic modifications of histones, recruitment of regulatory proteins to chromatin and changes to nuclear structure. To identify genes and regulatory elements responsive to synaptic activation in vivo, we performed a genome-wide ChIPseq analysis of acetylated histone H3 using somatosensory cortex of mice exposed to novel enriched environmental (NEE) conditions. We discovered that Short Interspersed Elements (SINEs) located distal to promoters of activity-dependent genes became acetylated following exposure to NEE and were bound by the general transcription factor TFIIIC. Importantly, under depolarizing conditions, inducible genes relocated to transcription factories (TFs), and this event was controlled by TFIIIC. Silencing of the TFIIIC subunit Gtf3c5 in non-stimulated neurons induced uncontrolled relocation to TFs and transcription of activity-dependent genes. Remarkably, in cortical neurons, silencing of Gtf3c5 mimicked the effects of chronic depolarization, inducing a dramatic increase of both dendritic length and branching. These findings reveal a novel and essential regulatory function of both SINEs and TFIIIC in mediating gene relocation and transcription. They also suggest that TFIIIC may regulate the rearrangement of nuclear architecture, allowing the coordinated expression of activity-dependent neuronal genes.
Synaptic long-term potentiation (LTP) is a key mechanism involved in learning and memory, and its alteration is associated with mental disorders. Shank3 is a major postsynaptic scaffolding protein that orchestrates dendritic spine morphogenesis, and mutations of this protein lead to mental retardation and autism spectrum disorders. In the present study we investigated the role of a new Shank3-associated protein in LTP. We identified the Rho-GAP interacting CIP4 homolog 2 (Rich2) as a new Shank3 partner by proteomic screen. Using single-cell bioluminescence resonance energy transfer microscopy, we found that Rich2-Shank3 interaction is increased in dendritic spines of mouse cultured hippocampal neurons during LTP. We further characterized Rich2 as an endosomal recycling protein that controls AMPA receptor GluA1 subunit exocytosis and spine morphology. Knock-down of Rich2 with siRNA, or disruption of the Rich2-Shank3 complex using an interfering mimetic peptide, inhibited the dendritic spine enlargement and the increase in GluA1 subunit exocytosis typical of LTP. These results identify Rich2-Shank3 as a new postsynaptic protein complex involved in synaptic plasticity.
The majority of neurons in the nervous system exhibit a polarized morphology, with multiple short dendrites and a single long axon. It is clear that multiple factors govern polarization in developing neurons, and the biased accumulation of intrinsic determinants to one side of the cell, coupled with responses to asymmetrically localized extrinsic factors, appears to be crucial. A number of intrinsic factors have been identified, but surprisingly little is known about the identity of the extrinsic signals. Here, we show in vivo that neuropilin-1 (Nrp1) and its co-receptor plexinA1 (Plxna1) are necessary to bias the extension of the dendrites of retinal ganglion cells to the apical side of the cell, and ectopically expressed class III semaphorins (Sema3s) disrupt this process. Importantly, the requirement for Nrp1 and Plxna1 in dendrite polarization occurs at a developmental time point after the cells have already extended their basally directed axon. Thus, we propose a novel mechanism whereby an extrinsic factor, probably a Sema3, acts through Nrp1 and Plxna1 to promote the asymmetric outgrowth of dendrites independently of axon polarization.
The tetra(ethylene glycol) derivative of benzothiazole aniline, BTA-EG4, is a novel amyloid-binding small molecule that can penetrate the blood-brain barrier and protect cells from Aβ-induced toxicity. However, the effects of Aβ-targeting molecules on other cellular processes, including those that modulate synaptic plasticity, remain unknown. We report here that BTA-EG4 decreases Aβ levels, alters cell surface expression of amyloid precursor protein (APP), and improves memory in wild-type mice. Interestingly, the BTA-EG4-mediated behavioral improvement is not correlated with LTP, but with increased spinogenesis. The higher dendritic spine density reflects an increase in the number of functional synapses as determined by increased miniature EPSC (mEPSC) frequency without changes in presynaptic parameters or postsynaptic mEPSC amplitude. Additionally, BTA-EG4 requires APP to regulate dendritic spine density through a Ras signaling-dependent mechanism. Thus, BTA-EG4 may provide broad therapeutic benefits for improving neuronal and cognitive function, and may have implications in neurodegenerative disease therapy.
Chronic lymphocytic leukemia (CLL) is the most frequent leukemia in adults. We have analyzed exome sequencing data from 127 individuals with CLL and Sanger sequencing data from 214 additional affected individuals, identifying recurrent somatic mutations in POT1 (encoding protection of telomeres 1) in 3.5% of the cases, with the frequency reaching 9% when only individuals without IGHV@ mutations were considered. POT1 encodes a component of the shelterin complex and is the first member of this telomeric structure found to be mutated in human cancer. Somatic mutation of POT1 primarily occurs in gene regions encoding the two oligonucleotide-/oligosaccharide-binding (OB) folds and affects key residues required to bind telomeric DNA. POT1-mutated CLL cells have numerous telomeric and chromosomal abnormalities that suggest that POT1 mutations favor the acquisition of the malignant features of CLL cells. The identification of POT1 as a new frequently mutated gene in CLL may facilitate novel approaches for the clinical management of this disease.
Intermediate filaments are cytoskeletal elements important for cell architecture. Recently it has been discovered that intermediate filaments are highly dynamic and that they are fundamental for organelle positioning, transport and function thus being an important regulatory component of membrane traffic. We have identified, using the yeast two-hybrid system, vimentin, a class III intermediate filament protein, as a Rab7a interacting protein. Rab7a is a member of the Rab family of small GTPases and it controls vesicular membrane traffic to late endosomes and lysosomes. In addition, Rab7a is important for maturation of phagosomes and autophagic vacuoles. We confirmed the interaction in HeLa cells by co-immunoprecipitation and pull-down experiments, and established that the interaction is direct using bacterially expressed recombinant proteins. Immunofluorescence analysis on HeLa cells indicate that Rab7a-positive vesicles sometimes overlap with vimentin filaments. Overexpression of Rab7a causes an increase in vimentin phosphorylation at different sites and causes redistribution of vimentin in the soluble fraction. Consistently, Rab7a silencing causes an increase of vimentin present in the insoluble fraction (assembled). Also, expression of Charcot-Marie-Tooth 2B-causing Rab7a mutant proteins induces vimentin phosphorylation and increases the amount of vimentin in the soluble fraction. Thus, modulation of expression levels of Rab7a wt or expression of Rab7a mutant proteins changes the assembly of vimentin and its phosphorylation state indicating that Rab7a is important for the regulation of vimentin function.
Laminins are large heterotrimeric cross-shaped extracellular matrix glycoproteins with terminal globular domains and a coiled-coil region through which the three chains are assembled and covalently linked. Laminins are key components of basement membranes, and they serve as attachment sites for cell adhesion, migration and proliferation. In this work, we produced a recombinant fragment comprising the entire laminin coiled-coil of the α1-, β1-, and γ1-chains that assemble into a stable heterotrimeric coiled-coil structure independently of the rest of the molecule. This domain was biologically active and not only failed to serve as a substrate for cell attachment, spreading and focal adhesion formation but also inhibited cell adhesion to laminin when added to cells in a soluble form at the time of seeding. Furthermore, gene array expression profiling in cells cultured in the presence of the laminin coiled-coil domain revealed up-regulation of genes involved in cell motility and invasion. These findings were confirmed by real-time quantitative PCR and zymography assays. In conclusion, this study shows for the first time that the laminin coiled-coil domain displays anti-adhesive functions and has potential implications for cell migration during matrix remodeling.
Multiple signaling pathways control the specification of endothelial cells (ECs) to become arteries or veins during vertebrate embryogenesis. Current models propose that a cascade of Hedgehog (Hh), vascular endothelial growth factor (VEGF), and Notch signaling acts instructively on ECs to control the choice between arterial or venous fate. Differences in the phenotypes induced by Hh, VEGF, or Notch inhibition suggest that not all of the effects of Hh on arteriovenous specification are mediated by VEGF. We establish that full derepression of the Hh pathway in ptc1;ptc2 mutants converts the posterior cardinal vein into a second arterial vessel that manifests intact arterial gene expression, intersegmental vessel sprouting, and HSC gene expression. Importantly, although VEGF was thought to be absolutely essential for arterial fates, we find that normal and ectopic arterial differentiation can occur without VEGF signaling in ptc1;ptc2 mutants. Furthermore, Hh is able to bypass VEGF to induce arterial differentiation in ECs via the calcitonin receptor-like receptor, thus revealing a surprising complexity in the interplay between Hh and VEGF signaling during arteriovenous specification. Finally, our experiments establish a dual function of Hh during induction of runx1(+) HSCs.
Cutaneous melanomas may be quite heterogeneous in their clinical, histological and molecular findings. Correlating these features may help identify distinctive subgroups of melanomas and improve our overall understanding and prognostication of melanoma. We recently identified a subgroup of melanomas with increased chromosomal copy number gains in 8q24 at MYC having several distinctive clinical and histopathological characteristics, including an aggressive clinical course and an amelanotic clinical and histological appearance. It has been postulated that oncogenes such as MYC may have regulatory effects on genes critical to melanin pigment synthesis, specifically microphthalmia-associated transcription factor (MITF), which is known to have a key role in regulating the expression of tyrosinase (TYR), an important enzyme in the production of melanin pigment. We investigated the possible mechanism underlying the amelanotic appearance of melanomas with gains in 8q24 by evaluating the relationship between melanomas with and without 8q24 copy number gains and c-MYC, MITF and TYR protein expression. Immunohistochemical analysis of c-MYC, MITF and TYR was performed on 36 melanomas with gains in 8q24 and 40 melanomas without gains in 8q24. The melanomas with gains of 8q24 correlated with elevated c-MYC protein expression and melanomas without gains in 8q24 showed significantly decreased c-MYC protein expression. A direct relationship between the presence of gains in 8q24 and decreased MITF expression, as well as between c-MYC and TYR protein expression was also observed. Our results suggest that MYC can have a role in the pigmentary pathway of melanoma. In amelanotic melanomas with gains in 8q24, downregulation of TYR and other melanocyte-specific genes may be mediated by MYC leading to transcriptional suppression of MITF. As MITF is a frequently used marker to establish melanocytic lineage in melanoma, our study also raises the important clinical consideration that amelanotic melanomas, especially those with gains in 8q24 may lack expression of MITF.
Notch signaling involves the proteolytic cleavage of the transmembrane Notch receptor after binding to its transmembrane ligands. Jagged-1 also undergoes proteolytic cleavage by gamma-secretase and releases an intracellular fragment. In this study, we have demonstrated that the Jagged-1 intracellular domain (JICD) inhibits Notch1 signaling via a reduction in the protein stability of the Notch1 intracellular domain (Notch1-IC). The formation of the Notch1-IC-RBP-Jk-Mastermind complex is prevented in the presence of JICD, via a physical interaction. Furthermore, JICD accelerates the protein degradation of Notch1-IC via Fbw7-dependent proteasomal pathway. These results indicate that JICD functions as a negative regulator in Notch1 signaling via the promotion of Notch1-IC degradation.
Homeodomain transcription factors classically exert their morphogenetic activities through the cell-autonomous regulation of developmental programs. In vertebrates, several homeoproteins have also been shown to have direct non-cell-autonomous activities in the developing nervous system. We present the first in vivo evidence for homeoprotein signaling in Drosophila. Focusing on wing development as a model, we first demonstrate that the homeoprotein Engrailed (En) is secreted. Using single-chain anti-En antibodies expressed under the control of a variety of promoters, we delineate the wing territories in which secreted En acts. We show that En is a short-range signaling molecule that participates in anterior crossvein development, interacting with the Dpp signaling pathway. This report thus suggests that direct signaling with homeoproteins is an evolutionarily conserved phenomenon that is not restricted to neural tissues and involves interactions with bona fide signal transduction pathways.
HP1 enrichment at pericentric heterochromatin is considered important for centromere function. Although HP1 binding to H3K9me3 can explain its accumulation at pericentric heterochromatin, how it is initially targeted there remains unclear. Here, in mouse cells, we reveal the presence of long nuclear noncoding transcripts corresponding to major satellite repeats at the periphery of pericentric heterochromatin. Furthermore, we find that major transcripts in the forward orientation specifically associate with SUMO-modified HP1 proteins. We identified this modification as SUMO-1 and mapped it in the hinge domain of HP1α. Notably, the hinge domain and its SUMOylation proved critical to promote the initial targeting of HP1α to pericentric domains using de novo localization assays, whereas they are dispensable for maintenance of HP1 domains. We propose that SUMO-HP1, through a specific association with major forward transcript, is guided at the pericentric heterochromatin domain to seed further HP1 localization.
Carbonic anhydrase IX (CAIX) is a hypoxia-induced, membrane-tethered enzyme that is highly expressed in many cancers. It catalyses the hydration of CO(2) to HCO(3)(-) and H(+), and the reverse dehydration reaction. Recent studies have shown an important role for CAIX in pH regulation and it has been speculated that CAIX may play a role in supporting cancer progression towards more aggressive forms of the disease. Clinical correlative studies in many tumours have shown that high expression is related to poor outcome. In the present study, we have selected antigen-binding antibody fragments (Fab) against human CAIX by phage-display, and tested these for inhibitory potency on CAIX catalytic activity. Inhibition was assessed from the kinetics of the CAIX-catalysed reaction, using assays performed on intact cells over-expressing CAIX, and their CAIX-containing membrane fragments. Inhibition was also assessed in multi-cellular tissue-models (spheroids) from the kinetics of CO(2) venting. We have identified a Fab antibody, labelled MSC8, and its corresponding full-length IgG that inhibited CAIX by up to 57% and 76%, respectively, with half-maximal inhibition at 0.3μg/ml. Incubation of CAIX-expressing cells with MSC8 IgG produced a lasting inhibitory effect. The inhibitory effect was prompt and was also observed in isolated membrane-fragments, suggesting that a direct inhibitory interaction takes place between the antibody and CAIX. The inhibitory effects in spheroids argue for a physiological relevance of the antibody. Biologically-active antibodies against CAIX can be used as selective, high-affinity inhibitors in experimental studies to dissect the role of CAIX and, possibly, therapeutically by targeting a catalytically-active cancer-related protein.
Extensins, hydroxyproline-rich repetitive glycoproteins with Ser-Hyp(4) motifs, are structural proteins in plant cell walls. The leucine-rich repeat extensin 1 (LRX1) of Arabidopsis thaliana is an extracellular protein with both a leucine-rich repeat and an extensin domain, and has been demonstrated to be important for cell-wall formation in root hairs. lrx1 mutants develop defective cell walls, resulting in a strong root hair phenotype. The extensin domain is essential for protein function and is thought to confer insolubilization of LRX1 in the cell wall. Here, in vivo characterization of the LRX1 extensin domain is described. First, a series of LRX1 extensin deletion constructs was produced that led to identification of a much shorter, functional extensin domain. Tyr residues can induce intra- and inter-molecular cross-links in extensins, and substitution of Tyr in the extensin domain by Phe led to reduced activity of the corresponding LRX1 protein. An additional function of Tyr (or Phe) is provided by the aromatic nature of the side chain. This suggests that these residues might be involved in hydrophobic stacking, possibly as a mechanism of protein assembly. Finally, modified LRX1 proteins lacking Tyr in the extensin domain are still insolubilized in the cell wall, indicating strong interactions of extensins within the cell wall in addition to the well-described Tyr cross-links.
Aristolochic acid (AA) is the causative agent of urothelial tumors associated with AA nephropathy and is also implicated in the development of Balkan endemic nephropathy-associated urothelial tumors. These tumors contain AA-characteristic TP53 mutations. We examined gene expression changes in Hupki (human TP53 knock-in) mice after treatment with aristolochic acid I (AAI) by gavage (5 mg/kg body weight). After 3, 12 and 21 days of treatment gene expression profiles were investigated using Agilent Whole Mouse 44K Genome Oligo Array. Expression profiles were significantly altered by AAI treatment in both target (kidney) and nontarget (liver) tissue. Renal pathology and DNA adduct analysis confirmed kidney as the target tissue of AAI-induced toxicity. Gene ontology for functional analysis revealed that processes related to apoptosis, cell cycle, stress response, immune system, inflammatory response and kidney development were altered in kidney. Canonical pathway analysis indicated Nfκb, aryl hydrocarbon receptor, Tp53 and cell cycle signaling as the most important pathways modulated in kidney. Expression of Nfκb1 and other Nfκb-target genes was confirmed by quantitative real-time PCR (qRT-PCR) and was consistent with the induction of Nfκb1 protein. Myc oncogene, frequently overexpressed in urothelial tumors, was upregulated by AAI on the microarrays and confirmed by qRT-PCR and protein induction. Collectively we found that microarray gene expression analysis is a useful tool to define tissue-specific responses in AAI-induced toxicity. Several genes identified such as TP53, Rb1, Mdm2, Cdkn2a and Myc are frequently affected in human urothelial cancer, and may be valuable prognostic markers in future clinical studies.
Mechanical forces play a crucial role in controlling the integrity and functionality of cells and tissues. External forces are sensed by cells and translated into signals that induce various responses. To increase the detailed understanding of these processes, we investigated cell migration and dynamic cellular reorganisation of focal adhesions and cytoskeleton upon application of cyclic stretching forces. Of particular interest was the role of microtubules and GTPase activation in the course of mechanotransduction. We showed that focal adhesions and the actin cytoskeleton undergo dramatic reorganisation perpendicular to the direction of stretching forces even without microtubules. Rather, we found that microtubule orientation is controlled by the actin cytoskeleton. Using biochemical assays and fluorescence resonance energy transfer (FRET) measurements, we revealed that Rac1 and Cdc42 activities did not change upon stretching, whereas overall RhoA activity increased dramatically, but independently of intact microtubules. In conclusion, we demonstrated that key players in force-induced cellular reorganisation are focal-adhesion sliding, RhoA activation and the actomyosin machinery. In contrast to the importance of microtubules in migration, the force-induced cellular reorganisation, including focal-adhesion sliding, is independent of a dynamic microtubule network. Consequently, the elementary molecular mechanism of cellular reorganisation during migration is different to the one in force-induced cell reorganisation.
The dendritic morphology of neurons dictates their abilities to process and transmit information; however, the signaling pathways that regulate dendritic growth and complexity are poorly understood. Here, we show that retinoids induce the expression of the FERM Rho-GEF protein FARP1 in the developing spinal cord. FARP1 is expressed in subsets of motor neurons and is enriched in dendrites of lateral motor column (LMC) neurons that innervate the limb. FARP1 is necessary and sufficient to promote LMC dendritic growth but does not affect dendrite number or axonal morphology. We show that FARP1 serves as a specific effector of transmembrane Semaphorin6A and PlexinA4 signals to regulate LMC dendritic growth, and that its Rho-GEF domain is necessary for this function. These findings reveal that retinoid and Sema6A/PlexA4 signaling pathways intersect through FARP1 to control dendritic growth, and uncover the existence of subtype-specific signaling networks that control dendritic developmental programs in spinal motor neurons.
Contrary to malignant melanoma, nevi are a benign form of melanocytic hyperproliferation. They are frequently observed as precursor lesions of melanoma, but they also feature biochemical markers of senescence. In particular, evidence for oncogene-induced melanocyte senescence as natural means to prevent tumorigenesis has been obtained in nevi with mutated B-Raf(V600E). Here, we demonstrate that strong oncogenic growth factor receptor signalling drives melanocytes into senescence, whereas weaker signals keep them in the proliferative state. Activation of oncogene-induced senescence also produces multinucleated giant cells, a long known histological feature of nevus cells. The protein levels of the senescence mediators, p53 and pRB, and their upstream activators do not correlate with senescence. However, strong oncogene signalling leads to pronounced reactive oxygen stress, and scavenging of reactive oxygen species (ROS) efficiently prevents the formation of multinucleated cells and senescence. Similarly, expression of oncogenic N-RAS results in ROS generation, DNA damage and the same multinuclear senescent phenotype. Hence, we identified oncogenic signalling-dependent ROS production as critical mediator of the melanocytic multinuclear phenotype and senescence, both of them being hallmarks of human nevus cells.
The GAGA factor (GAF), encoded by the Trithorax like gene (Trl) is a multifunctional protein involved in gene activation, Polycomb-dependent repression, chromatin remodeling and is a component of chromatin domain boundaries. Although first isolated as transcriptional activator of the Drosophila homeotic gene Ultrabithorax (Ubx), the molecular basis of this GAF activity is unknown. Here we show that dmTAF3 (also known as BIP2 and dTAF(II)155), a component of TFIID, interacts directly with GAF. We generated mutations in dmTAF3 and show that, in Trl mutant background, they affect transcription of Ubx leading to enhancement of Ubx phenotype. These results reveal that the gene activation pathway involving GAF is through its direct interaction with dmTAF3.
Charcot-Marie-Tooth (CMT) type 2 neuropathies are a group of autosomal-dominant axonal disorders genetically and clinically heterogeneous. In particular, CMT type 2B (CMT2B) neuropathies are characterized by severe sensory loss, often complicated by infections, arthropathy, and amputations. Recently, four missense mutations in the small GTPase Rab7 associated with the Charcot-Marie Tooth type 2B phenotype have been identified. These mutations target highly conserved amino acid residues. However, nothing is known about whether and how these mutations affect Rab7 function. We investigated the biochemical and functional properties of three of the mutant proteins. Interestingly, all three proteins exhibited higher nucleotide exchange rates and hydrolyzed GTP slower than the wild-type protein. In addition, whereas 23% of overexpressed wild-type Rab7 was GTP bound in HeLa cells, the large majority of the mutant proteins (82-89%) were in the GTP-bound form, consistent with the data on GTP hydrolysis and exchange rates. The CMT2B-associated Rab7 proteins were also able to bind the Rab7 effector RILP (Rab-interacting lysosomal protein) and to rescue Rab7 function after silencing. Altogether, these data demonstrate that all tested CMT2B-associated Rab7 mutations are mechanistically similar, suggesting that activated forms of the Rab7 are responsible for CMT2B disease.
The liver peptide hepcidin regulates iron absorption and recycling. Hemojuvelin (HJV) has a key role in hepcidin regulation, and its inactivation causes severe iron overload both in humans and in mice. Membrane HJV (m-HJV) acts as a coreceptor for bone morphogenetic proteins (BMPs), whereas soluble HJV (s-HJV) may down-regulate hepcidin in a competitive way interfering with BMP signaling. s-HJV is decreased by iron in vitro and increased by iron deficiency in vivo. However, the mechanisms regulating the 2 HJV isoforms remain unclear. Here we show that s-HJV originates from a furin cleavage at position 332-335. s-HJV is reduced in the cleavage mutant R335Q as well as in cells treated with a furin inhibitor, and increased in cells overexpressing exogenous furin, but not in cells overexpressing an inactive furin variant. Furin is up-regulated by iron deficiency and hypoxia in association with the stabilization of HIF-1alpha. Increased s-HJV in response to HIF-1alpha occurs during differentiation of murine muscle cells expressing endogenous Hjv. Our data are relevant to the mechanisms that relate iron metabolism to the hypoxic response. The release of s-HJV might be a tissue-specific mechanism, signaling the local iron requests of hypoxic skeletal muscles independently of the oxygen status of the liver.
Recently, we demonstrated that a polymorphism in exon 2 of the serum carnosinase (CNDP1) gene is associated with susceptibility to developing diabetic nephropathy. Based on the number of CTG repeats in the signal peptide, five different alleles coding for 4, 5, 6, 7, or 8 leucines (4L-8L) are known. Diabetic patients without nephropathy are homozygous for the 5L allele more frequently than those with nephropathy. Since serum carnosinase activity correlates with CNDP1 genotype, we hypothesized in the present study that secretion of serum carnosinase is determined by the CNDP1 genotype. To test this hypothesis, we transfected Cos-7 cells with different CNDP1 constructs varying in CTG repeats and assessed the expression of CNDP1 protein in cell extracts and supernatants. Our results demonstrate that CNDP1 secretion is significantly higher in cells expressing variants with more than five leucines in the signal peptide. Hence, our data might explain why individuals homozygous for the 5L allele have low serum carnosinase activity. Because carnosine, the natural substrate for carnosinase, exerts antioxidative effects and inhibits ACE activity and advanced glycation end product formation, our results support the finding that diabetic patients homozygous for CNDP1 5L are protected against diabetic nephropathy.
Cell shape changes require the coordination of actin and microtubule cytoskeletons. The molecular mechanisms by which such coordination is achieved remain obscure, particularly in the context of epithelial cells within developing vertebrate embryos. We have identified a novel role for the actin-binding protein Shroom3 as a regulator of the microtubule cytoskeleton during epithelial morphogenesis. We show that Shroom3 is sufficient and also necessary to induce a redistribution of the microtubule regulator gamma-tubulin. Moreover, this change in gamma-tubulin distribution underlies the assembly of aligned arrays of microtubules that drive apicobasal cell elongation. Finally, experiments with the related protein, Shroom1, demonstrate that gamma-tubulin regulation is a conserved feature of this protein family. Together, the data demonstrate that Shroom family proteins govern epithelial cell behaviors by coordinating the assembly of both microtubule and actin cytoskeletons.
Excitotoxicity mediated by glutamate receptors plays crucial roles in ischemia and other neurodegenerative diseases. Whereas overactivation of ionotropic glutamate receptors is neurotoxic, the role of metabotropic glutamate receptors (mGluRs), and especially mGluR1, remains equivocal. Here we report that activation of NMDA receptors results in calpain-mediated truncation of the C-terminal domain of mGluR1alpha at Ser(936). The truncated mGluR1alpha maintains its ability to increase cytosolic calcium while it no longer activates the neuroprotective PI(3)K-Akt signaling pathways. Full-length and truncated forms of mGluR1alpha play distinct roles in excitotoxic neuronal degeneration in cultured neurons. A fusion peptide derived from the calpain cleavage site of mGluR1alpha efficiently blocks NMDA-induced truncation of mGluR1alpha in primary neuronal cultures and exhibits neuroprotection against excitotoxicity both in vitro and in vivo. These findings shed light on the relationship between NMDA and mGluR1alpha and indicate the existence of a positive feedback regulation in excitotoxicity involving calpain and mGluR1alpha.
Protein phosphatase 1 (PP1) is a ubiquitous serine/threonine phosphatase that regulates many cellular processes, including cell division, signaling, differentiation, and metabolism. It is expressed in mammalian cells as three closely related isoforms: alpha, beta/delta, and gamma1. These isoforms differ in their relative affinities for proteins, termed targeting subunits, that mediate their intracellular localization and substrate specificity. Because of the dynamic nature of these interactions, it is important to find experimental approaches that permit direct analyses of PP1 localization and PP1-targeting subunit interactions in live cells. When transiently or stably expressed as fluorescent protein (FP) fusions, the three isoforms are active phosphatases with distinct localization patterns and can interact with both endogenous and exogenous targeting subunits. Their changing spatio-temporal distributions can be monitored both throughout the cell cycle and following cellular perturbations by time-lapse fluorescence microscopy, and turnover rates of intracellular pools of the protein calculated by fluorescence recovery after photobleaching (FRAP). Interactions with targeting subunits can be visualized in vivo by fluorescence resonance energy transfer (FRET), using techniques such as sensitized emission, acceptor photobleaching, or fluorescence lifetime imaging.
Although enhanced cardiac matrix metalloproteinase (MMP)-2 synthesis has been associated with ventricular remodeling and failure, whether MMP-2 expression is a direct mediator of this process is unknown. We generated transgenic mice expressing active MMP-2 driven by the alpha-myosin heavy chain promoter. At 4 mo MMP-2 transgenic hearts demonstrated expression of the MMP-2 transgene, myocyte hypertrophy, breakdown of Z-band registration, lysis of myofilaments, disruption of sarcomere and mitochondrial architecture, and cardiac fibroblast proliferation. Hearts from 8-mo-old transgenic mice displayed extensive myocyte disorganization and dropout with replacement fibrosis and perivascular fibrosis. Older transgenic mice also exhibited a massive increase in cardiac MMP-2 expression, representing recruitment of endogenous MMP-2 synthesis, with associated expression of MMP-9 and membrane type 1 MMP. Increases in diastolic [control (C) 33 +/- 3 vs. MMP 51 +/- 12 microl; P = 0.003] and systolic (C 7 +/- 2 vs. MMP 28 +/- 14 microl; P = 0.003) left ventricular (LV) volumes and relatively preserved stroke volume (C 26 +/- 4 vs. MMP 23 +/- 3 microl; P = 0.16) resulted in markedly decreased LV ejection fraction (C 78 +/- 7% vs. MMP 48 +/- 16%; P = 0.0006). Markedly impaired systolic function in the MMP transgenic mice was demonstrated in the reduced preload-adjusted maximal power (C 240 +/- 84 vs. MMP 78 +/- 49 mW/microl(2); P = 0.0003) and decreased end-systolic pressure-volume relation (C 7.5 +/- 1.5 vs. MMP 4.7 +/- 2.0; P = 0.016). Expression of active MMP-2 is sufficient to induce severe ventricular remodeling and systolic dysfunction in the absence of superimposed injury.
Histone posttranslational modifications (PTMs) and sequence variants regulate genome function. Although accumulating evidence links particular PTM patterns with specific genomic loci, our knowledge concerning where and when these PTMs are imposed remains limited. Here, we find that lysine methylation is absent prior to histone incorporation into chromatin, except at H3K9. Nonnucleosomal H3.1 and H3.3 show distinct enrichments in H3K9me, such that H3.1 contains more K9me1 than H3.3. In addition, H3.3 presents other modifications, including K9/K14 diacetylated and K9me2. Importantly, H3K9me3 was undetectable in both nonnucleosomal variants. Notably, initial modifications on H3 variants can potentiate the action of enzymes as exemplified with Suv39HMTase to produce H3K9me3 as found in pericentric heterochromatin. Although the set of initial modifications present on H3.1 is permissive for further modifications, in H3.3 a subset cannot be K9me3. Thus, initial modifications impact final PTMs within chromatin.
Loc1p is an exclusively nuclear dsRNA-binding protein that affects the asymmetric sorting of ASH1 mRNA to daughter cells in Saccharomyces cerevisiae. In addition to the role in cytoplasmic RNA localization, Loc1p is a constituent of pre-60S ribosomes. Cells devoid of Loc1p display a defect in the synthesis of 60S ribosomal subunits, resulting in "half-mer" polyribosomes. Previously, we reported that Loc1p is located throughout the entire nucleus; however, upon closer inspection we discovered that Loc1p is enriched in the nucleolus consistent with a role in 60S ribosome biogenesis. Given that Loc1p is an RNA-binding protein and presumably functions in the assembly of 60S ribosomal subunits, we investigated if Loc1p has a role in rRNA processing and nuclear export of 60S subunits. Analysis of pre-rRNA processing revealed that loc1Delta cells exhibit gross defects in 25S rRNA synthesis, specifically a delay in processing at sites A0, A1 and A2 in 35S pre-rRNA. Furthermore, loc1Delta cells exhibit nuclear export defects for 60S ribosomal subunits, again, consistent with a role for Loc1p in the assembly of 60S ribosomal subunits. It is attractive to hypothesize that the two phenotypes associated with loc1Delta cells, namely altered ASH1 mRNA localization and ribosome biogenesis, are not mutually exclusive, but that ribosome biogenesis directly impacts mRNA localization.
Healthy volunteers are hyperimmunized with RhD-positive red cells in order to obtain plasma containing high titres of anti-D immunoglobulin, which is used for the prevention of haemolytic disease of the fetus and newborn. We analysed the anti-D immune response in a donor who had been hyperimmunized for 7 years and who showed declining anti-D titres despite re-immunization. A phage display library representing the complete immunorepertoire and a second library representing the IGHV3 superspecies family genes (IGHV3s) repertoire in the donor were constructed and analysed. A clonal Ig-gene rearrangement was quantified in the peripheral blood by limiting dilution polymerase chain reaction (PCR) All RhD-binding phages from both libraries, except one, had heavy chains with IGH-VDJ rearrangements of the same clonal origin, but with different patterns of somatic mutations and joined with different light chains. Limiting dilution PCR performed on mRNA and genomic DNA showed a frequency of 1 clonal B cell in 2000 IgG1/3-positive B cells. We show the presence of clonally related RhD-specific B cells in a hyperimmunized anti-D donor who had declining anti-D titres and who was unresponsive to re-immunization. Furthermore, we found a high frequency of clonal B cells. These results contribute to the understanding of the immune response against RhD in hyperimmunized anti-D donors.
The anaphase-promoting complex (APC) or cyclosome is a multi-subunit ubiquitin ligase that controls progression through mitosis and the G1-phase of the cell cycle. The APC ubiquitinates regulatory proteins such as securin and cyclin B and thereby targets them for destruction by the 26S proteasome. Activation of the APC depends on the activator proteins Cdc20 and Cdh1, which are thought to recruit substrates to the APC. In vitro, APC's RING finger subunit Apc11 alone can also function as a ubiquitin ligase. Here, we review different methods that have been used to measure the ubiquitination activity of the APC in vitro and to analyze APC-mediated degradation reactions either in vitro or in vivo. We describe procedures to isolate the APC from human cells or from Xenopus eggs, to activate purified APC with recombinant Cdc20 or Cdh1 and to measure the ubiquitination activity of the resulting APC(Cdc20) and APC(Cdh1) complexes. We also describe procedures to analyze the ubiquitination activity associated with recombinant Apc11.
One of the most clinically advanced forms of experimental disease-modifying treatment for Alzheimer disease is immunization against the amyloid beta protein (Abeta), but how this may prevent cognitive impairment is unclear. We hypothesized that antibodies to Abeta could exert a beneficial action by directly neutralizing potentially synaptotoxic soluble Abeta species in the brain. Intracerebroventricular injection of naturally secreted human Abeta inhibited long-term potentiation (LTP), a correlate of learning and memory, in rat hippocampus in vivo but a monoclonal antibody to Abeta completely prevented the inhibition of LTP when injected after Abeta. Size fractionation showed that Abeta oligomers, not monomers or fibrils, were responsible for inhibiting LTP, and an Abeta antibody again prevented such inhibition. Active immunization against Abeta was partially effective, and the effects correlated positively with levels of antibodies to Abeta oligomers. The ability of exogenous and endogenous antibodies to rapidly neutralize soluble Abeta oligomers that disrupt synaptic plasticity in vivo suggests that treatment with such antibodies might show reversible cognitive deficits in early Alzheimer disease.
Individuals with permanent neonatal diabetes mellitus usually present within the first three months of life and require insulin treatment. We recently identified a locus on chromosome 10p13-p12.1 involved in permanent neonatal diabetes mellitus associated with pancreatic and cerebellar agenesis in a genome-wide linkage search of a consanguineous Pakistani family. Here we report the further linkage analysis of this family and a second family of Northern European descent segregating an identical phenotype. Positional cloning identified the mutations 705insG and C886T in the gene PTF1A, encoding pancreas transcription factor 1alpha, as disease-causing sequence changes. Both mutations cause truncation of the expressed PTF1A protein C-terminal to the basic-helix-loop-helix domain. Reporter-gene studies using a minimal PTF1A deletion mutant indicate that the deleted region defines a new domain that is crucial for the function of this protein. PTF1A is known to have a role in mammalian pancreatic development, and the clinical phenotype of the affected individuals implicated the protein as a key regulator of cerebellar neurogenesis. The essential role of PTF1A in normal cerebellar development was confirmed by detailed neuropathological analysis of Ptf1a(-/-) mice.
Imprinted genes are expressed from only one of the parental alleles and are marked epigenetically by DNA methylation and histone modifications. The paternally expressed gene insulin-like growth-factor 2 (Igf2) is separated by approximately 100 kb from the maternally expressed noncoding gene H19 on mouse distal chromosome 7. Differentially methylated regions in Igf2 and H19 contain chromatin boundaries, silencers and activators and regulate the reciprocal expression of the two genes in a methylation-sensitive manner by allowing them exclusive access to a shared set of enhancers. Various chromatin models have been proposed that separate Igf2 and H19 into active and silent domains. Here we used a GAL4 knock-in approach as well as the chromosome conformation capture technique to show that the differentially methylated regions in the imprinted genes Igf2 and H19 interact in mice. These interactions are epigenetically regulated and partition maternal and paternal chromatin into distinct loops. This generates a simple epigenetic switch for Igf2 through which it moves between an active and a silent chromatin domain.
The 9E10 antibody epitope (EQKLISEEDL) derives from a protein sequence in the human proto-oncogen p62(c-myc) and is widely used as a protein fusion tag. This myc-tag is a powerful tool in protein localization, immunochemistry, ELISA or protein purification. Here, we characterize the myc-tag epitope by substitutional analysis and length variation using peptide spot synthesis on cellulose. The key amino acids of this interaction are the core residues LISE. The shortest peptide with a strong binding signal is KLISEEDL. Dissociation constants of selected peptide variants to the antibody 9E10 were determined. scFv constructs with the shortest possible myc-tags were successfully detected by Western blot and ELISA, giving a signal comparable to that of the original myc-tag.
Constitutive c-myc expression suppresses cell cycle arrest, promotes entry into S phase, and results in the growth factor-independent expression of ornithine decarboxylase (ODC; EC 4.1.1.17). The ODC gene contains a conserved repeat of the Myc binding site, CACGTG, in intron 1. In this report, we demonstrate that c-Myc is a potent transactivator of ODC promoter-reporter gene constructs in fibroblasts that requires the CACGTG repeat. These sites conferred Myc responsiveness on heterologous promoter constructs, suggesting that ODC is regulated by Myc at the level of transcription initiation. Analysis of deletion and point mutants of c-myc revealed that domains required for transactivation of the ODC promoter did not include the leucine zipper of the Myc protein. This suggests that Myc may interact with transcription factors other than Max to transactivate the ODC gene.
Three monoclonal antibodies were characterized by examining their reactivity to human cytomegalovirus (HCMV) glycoproteins under reducing and nonreducing conditions and their reactivity to glycoproteins and disulfide-linked glycoprotein complexes isolated by ion-exchange high-performance liquid chromatography. One monoclonal antibody, 9E10, reacted with glycoprotein complexes which had molecular weights of 93,000 and 450,000 and eluted from the ion-exchange column at 0.3 and 0.9 M NaCl, respectively. All glycoproteins associated in these complexes could be immunoprecipitated under reducing conditions by 9E10, suggesting that they were related to one another. The most abundant glycoproteins immunoprecipitated by 9E10 had molecular weights of 50,000 to 52,000. In contrast to this antibody, two other monoclonal antibodies, 9B7 and 41C2, reacted with glycoprotein complexes which had molecular weights of 130,000 and greater than 200,000 and eluted from the ion-exchange column at 0.6 M NaCl. All glycoproteins associated in these complexes could be immunoprecipitated by 9B7 or 41C2 under reducing conditions, suggesting that they were also related to one another. The most abundant glycoprotein immunoprecipitated by 41C2 or 9B7 had a molecular weight of 93,000. In addition, it was also determined that a 93,000-molecular-weight glycoprotein which was not associated with other glycoproteins by disulfide bonds could not be precipitated by any of the three antibodies, suggesting that it was different from the other glycoproteins. The monoclonal antibodies were also examined for specificity and neutralizing activity. Monoclonal antibodies 41C2 and 9B7 were specific to HCMV as determined by immunofluorescent staining of skin fibroblast cells infected with several different viruses. However, 41C2 did not neutralize Towne strain HCMV, while 9B7 did. The neutralizing activity of 9B7 did require complement. These results suggested that 41C2 and 9B7 reacted with different antigenic sites on the same glycoproteins. Unlike 41C2 and 9B7, monoclonal antibody 9E10 was found to cross-react with adenovirus and herpes simplex virus as determined by immunofluorescent staining of infected skin fibroblast cells. Furthermore, 9E10 neutralized the Towne and Toledo strains of HCMV in the absence of complement.
BACKGROUND & AIMS:
Activation of WNT signaling promotes the invasive activities of several types of cancer cells, but it is not clear if it regulates the same processes in colorectal cancer (CRC) cells, or what mechanisms are involved. We studied the expression and function of OVOL2, a member of the Ovo family of conserved zinc-finger transcription factors regulated by the WNT signaling pathway, in intestinal tumors of mice and human beings.
METHODS:
We analyzed the expression of OVOL2 protein and messenger RNA in CRC cell lines and tissue arrays, as well as CRC samples from patients who underwent surgery at Xiamen University in China from 2009 to 2012; clinical information also was collected. CRC cell lines (SW620) were infected with lentivirus expressing OVOL2, analyzed in migration and invasion assays, and injected into nude mice to assess tumor growth and metastasis. Tandem affinity purification was used to purify the OVOL2-containing complex from CRC cells; the complex was analyzed by liquid chromatography, tandem mass spectrometry, and immunoprecipitation experiments. Gene promoter activities were measured in luciferase reporter assays. We analyzed mice with an intestine-specific disruption of Ovol2 (Ovol2(flox/+) transgenic mice), as well as Apc(min/+) mice; these mice were crossed and analyzed.
RESULTS:
Analysis of data from patients indicated that the levels of OVOL2 messenger RNA were significantly lower in colon carcinomas than adenomas, and decreased significantly as carcinomas progressed from grades 2 to 4. Immunohistochemical analysis of a tissue array of 275 CRC samples showed a negative association between tumor stage and OVOL2 level. Overexpression of OVOL2 in SW620 cells decreased their migration and invasion, reduced markers of the epithelial-to-mesenchymal transition, and suppressed their metastasis as xenograft tumors in nude mice; knockdown of OVOL2 caused LS174T cells to transition from epithelial to mesenchymal phenotypes. OVOL2 bound T-cell factor (TCF)4 and β-catenin, facilitating recruitment of histone deacetylase 1 to the TCF4-β-catenin complex; this inhibited expression of epithelial-to-mesenchymal transition-related genes regulated by WNT, such as SLUG, in CRC cell lines. OVOL2 was a downstream target of WNT signaling in LS174T and SW480 cells. The OVOL2 promoter was hypermethylated in late-stage CRC specimens from patients and in SW620 cells; hypermethylation resulted in OVOL2 down-regulation and an inability to inhibit WNT signaling. Disruption of Ovol2 in Apc(min/+) mice increased WNT activity in intestinal tissues and the formation of invasive intestinal tumors.
CONCLUSIONS:
OVOL2 is a colorectal tumor suppressor that blocks WNT signaling by facilitating the recruitment of histone deacetylase 1 to the TCF4-β-catenin complex. Strategies to increase levels of OVOL2 might be developed to reduce colorectal tumor progression and metastasis.
Copyright © 2016 AGA Institute. Published by Elsevier Inc. All rights reserved.
OBJECTIVE:
Oncofetal protein insulin-like growth factor II mRNA-binding protein 1 (IMP1) regulates cellular proliferation and migration. Expression of IMP1 is limited to a few adult human tissues. However, it commonly expresses in a variety of cancers. Our objective was to study the regulatory mechanism of IMP1 on the cellular functions of choriocarcinoma (CC) JAR cells.
METHODS:
IMP1 protein levels were measured in CC tissues via immunohistochemistry. Specific siRNAs were used to down-regulate gene expressions. The abilities of migration and invasion were estimated by wound-healing and Matrigel chamber assays. The profile of IMP1-binding genes was investigated with an Agilent microarray. RT-qPCR, RNA immunoprecipitation, and IMP1 rescue experiments were performed to confirm the association between IMP1 and its binding genes. Gene expression was further analyzed by using RT-PCR and Western blotting.
RESULTS:
Strong IMP1 expressions were frequently detected in CC tissues. Knockdown of IMP1 expression in JAR cells inhibited cell migration and invasion, but did not affect cellular proliferation and morphology. Microarray and RNA-immunoprecipitation results revealed several candidate genes regulated by IMP1. Among them, ribosomal protein S6 kinase (RSK2) and protein phosphatase methylesterase 1 (PPME1) were confirmed to be down-regulated in IMP1-depleted JAR cells. Re-expression of IMP1 into the cells restored the expressions of RSK2 and PPME1. Furthermore, the depletion of RSK2 or PPME1 decreased the migration and invasion of JAR cells.
CONCLUSION:
Our results suggest that IMP1 plays an essential role in the regulation of migration and invasion of human CC cells, possibly through the novel effectors RSK2 and PPME1.
© 2013.
BACKGROUND:
Deregulation of the activity of the ubiquitin ligase E6AP (UBE3A) is well recognised to contribute to the development of Angelman syndrome (AS). The ubiquitin ligase HERC2, encoded by the HERC2 gene is thought to be a key regulator of E6AP.
METHODS AND RESULTS:
Using a combination of autozygosity mapping and linkage analysis, we studied an autosomal-recessive neurodevelopmental disorder with some phenotypic similarities to AS, found among the Old Order Amish. Our molecular investigation identified a mutation in HERC2 associated with the disease phenotype. We establish that the encoded mutant HERC2 protein has a reduced half-life compared with its wild-type counterpart, which is associated with a significant reduction in HERC2 levels in affected individuals.
CONCLUSIONS:
Our data implicate a model in which disruption of HERC2 function relates to a reduction in E6AP activity resulting in neurodevelopmental delay, suggesting a previously unrecognised role of HERC2 in the pathogenesis of AS.
AIM:
To understand the role of iPS inductive genes in esophageal cancer, we examined the expression of Sex determining region Y-box 2 (SOX2), Octamer-binding transcription factor 3/4 (OCT3/4), Krueppel-like factor 4 (KLF4), c-Myelocytomatosis viral oncogene (c-MYC) and Tir Na Nog (NANOG) using an esophageal squamous cell carcinoma tissue micrroarray.
MATERIALS AND METHODS:
The immunohistochemical expression levels of the five genes were compared to the clinicopathological data of the 81 patients with esophageal cancer.
RESULTS:
There was no relationship between the expression of the five genes and TNM factors of the patients. High expression of NANOG was an independent favorable prognostic factor (p=0.041). Among the patients who received postoperative cisplatin-based chemotherapy, patients with NANOG-positive tumor had significantly better prognosis than those whose tumors were NANOG negative (p=0.024). On the other hand, those with c-MYC-positive expression tended to have a worse prognosis and were resistant to cisplatin-based chemotherapy.
CONCLUSION:
NANOG expression was found to be an independent prognostic factor for patient with esophageal cancer. Patients with NANOG-positive expression tumor may be good candidates for cisplatin-based treatment.































































































































































































































































![Western Blot - Anti-Myc Tag Antibody [ARC5004-12] (A305783) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305783_1.jpg?profile=product_alternative)