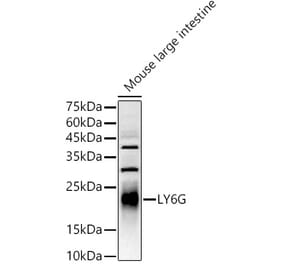
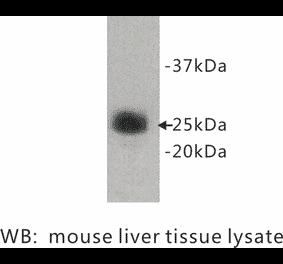
The rat monoclonal antibody RB6-8C5 detects Ly6G component of Gr-1 antigen, a commonly used surface marker of neutrophils.
Applications
FC, IP, WB, IHC-P, IHC-Fr, FUNC
Reactivity
Mouse
Immunogen
Murine granulocytes.
Host
Rat
Clonality
Monoclonal
Clone ID
RB6-8C5
Isotype
IgG2b
Conjugate
Unconjugated
Purification
Protein G chromatography.
Concentration
1 mg/ml
Purity
> 95% (by SDS-PAGE).
Product Form
Liquid
Formulation
Supplied in Phosphate Buffered Saline, pH 7.4, without Azide (0.2 µm filter sterilised). Endotoxin level is less than 0.01 EU/µg of the protein, as determined by the LAL test.