Unconjugated
Macrophages specialize in removing lipids and debris present in the atherosclerotic plaque. However, plaque progression renders macrophages unable to degrade exogenous atherogenic material and endogenous cargo including dysfunctional proteins and organelles. Here we show that a decline in the autophagy-lysosome system contributes to this as evidenced by a derangement in key autophagy markers in both mouse and human atherosclerotic plaques. By augmenting macrophage TFEB, the master transcriptional regulator of autophagy-lysosomal biogenesis, we can reverse the autophagy dysfunction of plaques, enhance aggrephagy of p62-enriched protein aggregates and blunt macrophage apoptosis and pro-inflammatory IL-1ß levels, leading to reduced atherosclerosis. In order to harness this degradative response therapeutically, we also describe a natural sugar called trehalose as an inducer of macrophage autophagy-lysosomal biogenesis and show trehalose's ability to recapitulate the atheroprotective properties of macrophage TFEB overexpression. Our data support this practical method of enhancing the degradative capacity of macrophages as a therapy for atherosclerotic vascular disease.
Damaged mitochondria pose a lethal threat to cells that necessitates their prompt removal. The currently recognized mechanism for disposal of mitochondria is autophagy, where damaged organelles are marked for disposal via ubiquitylation by Parkin. Here we report a novel pathway for mitochondrial elimination, in which these organelles undergo Parkin-dependent sequestration into Rab5-positive early endosomes via the ESCRT machinery. Following maturation, these endosomes deliver mitochondria to lysosomes for degradation. Although this endosomal pathway is activated by stressors that also activate mitochondrial autophagy, endosomal-mediated mitochondrial clearance is initiated before autophagy. The autophagy protein Beclin1 regulates activation of Rab5 and endosomal-mediated degradation of mitochondria, suggesting cross-talk between these two pathways. Abrogation of Rab5 function and the endosomal pathway results in the accumulation of stressed mitochondria and increases susceptibility to cell death in embryonic fibroblasts and cardiac myocytes. These data reveal a new mechanism for mitochondrial quality control mediated by Rab5 and early endosomes.
Whether obesity accelerates or suppresses autophagy in adipose tissue is still debatable. To clarify dysregulation of autophagy and its role in pathologies of obese adipose tissue, we focused on lysosomal function, protease maturation and activity, both in vivo and in vitro. First, we showed that autophagosome formation was accelerated, but autophagic clearance was impaired in obese adipose tissue. We also found protein and activity levels of CTSL (cathepsin L) were suppressed in obese adipose tissue, while the activity of CTSB (cathepsin B) was significantly enhanced. Moreover, cellular senescence and inflammasomes were activated in obese adipose tissue. In 3T3L1 adipocytes, downregulation of CTSL deteriorated autophagic clearance, upregulated expression of CTSB, promoted cellular senescence and activated inflammasomes. Upregulation of CTSB promoted additional activation of inflammasomes. Therefore, we suggest lysosomal dysfunction observed in obese adipose tissue leads to lower autophagic clearance, resulting in autophagosome accumulation. Simultaneously, lysosomal abnormalities, including deteriorated CTSL function and compensatory activation of CTSB, caused cellular senescence and inflammasome activation. Our findings strongly suggest lysosomal dysfunction is involved in early pathologies of obese adipose tissue.
Membrane-bound solute carriers (SLCs) are essential as they maintain several physiological functions, such as nutrient uptake, ion transport and waste removal. The SLC family comprise about 400 transporters, and we have identified two new putative family members, major facilitator superfamily domain containing 1 (MFSD1) and 3 (MFSD3). They cluster phylogenetically with SLCs of MFS type, and both proteins are conserved in chordates, while MFSD1 is also found in fruit fly. Based on homology modelling, we predict 12 transmembrane regions, a common feature for MFS transporters. The genes are expressed in abundance in mice, with specific protein staining along the plasma membrane in neurons. Depriving mouse embryonic primary cortex cells of amino acids resulted in upregulation of Mfsd1, whereas Mfsd3 is unaltered. Furthermore, in vivo, Mfsd1 and Mfsd3 are downregulated in anterior brain sections in mice subjected to starvation, while upregulated specifically in brainstem. Mfsd3 is also attenuated in cerebellum after starvation. In mice raised on high-fat diet, Mfsd1 was specifically downregulated in brainstem and hypothalamus, while Mfsd3 was reduced consistently throughout the brain.
Adaptation to changes in nutrient availability is crucial for cells and organisms. Posttranslational modifications of signaling proteins are very dynamic and are therefore key to promptly respond to nutrient deprivation or overload. Herein we screened for ubiquitylation of proteins in the livers of fasted and refed mice using a comprehensive systemic proteomic approach. Among 1641 identified proteins, 117 were differentially ubiquitylated upon fasting or refeeding. Endoplasmic reticulum (ER) and secretory proteins were enriched in the livers of refed mice in part owing to an ER-stress-mediated response engaging retro-translocation and ubiquitylation of proteins from the ER. Complement C3, an innate immune factor, emerged as the most prominent ER-related hit of our screen. Accordingly, we found that secretion of C3 from the liver and primary hepatocytes as well as its dynamic trafficking are nutrient dependent. Finally, obese mice with a chronic nutrient overload show constitutive trafficking of C3 in the livers despite acute changes in nutrition, which goes in line with increased C3 levels and low-grade inflammation reported for obese patients. Our study thus suggests that nutrient sensing in the liver is coupled to release of C3 and potentially its metabolic and inflammatory functions.
Mucopolysaccharidosis type II (MPSII) is an X-linked lysosomal storage disease characterized by severe neurologic and somatic disease caused by deficiency of iduronate-2-sulfatase (IDS), an enzyme that catabolizes the glycosaminoglycans heparan and dermatan sulphate. Intravenous enzyme replacement therapy (ERT) currently constitutes the only approved therapeutic option for MPSII. However, the inability of recombinant IDS to efficiently cross the blood-brain barrier (BBB) limits ERT efficacy in treating neurological symptoms. Here, we report a gene therapy approach for MPSII through direct delivery of vectors to the CNS. Through a minimally invasive procedure, we administered adeno-associated virus vectors encoding IDS (AAV9-Ids) to the cerebrospinal fluid of MPSII mice with already established disease. Treated mice showed a significant increase in IDS activity throughout the encephalon, with full resolution of lysosomal storage lesions, reversal of lysosomal dysfunction, normalization of brain transcriptomic signature, and disappearance of neuroinflammation. Moreover, our vector also transduced the liver, providing a peripheral source of therapeutic protein that corrected storage pathology in visceral organs, with evidence of cross-correction of nontransduced organs by circulating enzyme. Importantly, AAV9-Ids-treated MPSII mice showed normalization of behavioral deficits and considerably prolonged survival. These results provide a strong proof of concept for the clinical translation of our approach for the treatment of Hunter syndrome patients with cognitive impairment.
Homo and heterozygote cx3cr1 mutant mice, which harbor a green fluorescent protein (EGFP) in their cx3cr1 loci, represent a widely used animal model to study microglia and peripheral myeloid cells. Here we report that microglia in the dentate gyrus (DG) of cx3cr1 (-/-) mice displayed elevated microglial sirtuin 1 (SIRT1) expression levels and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) p65 activation, despite unaltered morphology when compared to cx3cr1 (+/-) or cx3cr1 (+/+) controls. This phenotype was restricted to the DG and accompanied by reduced adult neurogenesis in cx3cr1 (-/-) mice. Remarkably, adult neurogenesis was not affected by the lack of the CX3CR1-ligand, fractalkine (CX3CL1). Mechanistically, pharmacological activation of SIRT1 improved adult neurogenesis in the DG together with an enhanced performance of cx3cr1 (-/-) mice in a hippocampus-dependent learning and memory task. The reverse condition was induced when SIRT1 was inhibited in cx3cr1 (-/-) mice, causing reduced adult neurogenesis and lowered hippocampal cognitive abilities. In conclusion, our data indicate that deletion of CX3CR1 from microglia under resting conditions modifies brain areas with elevated cellular turnover independent of CX3CL1.
Cardiovascular disease (CVD) is the leading cause of the death worldwide. An increasing number of studies have found that autophagy is involved in the progression or prevention of CVD. However, the precise mechanism of autophagy in CVD, especially the myocardial ischaemia-reperfusion injury (MI/R injury), is unclear and controversial. Here, we show that the cardiomyocyte-specific disruption of autophagy by conditional knockout of Atg7 leads to severe contractile dysfunction, myofibrillar disarray and vacuolar cardiomyocytes. A negative cytoskeleton organization regulator, CLP36, was found to be accumulated in Atg7-deficient cardiomyocytes. The cardiomyocyte-specific knockout of Atg7 aggravates the MI/R injury with cardiac hypertrophy, contractile dysfunction, myofibrillar disarray and severe cardiac fibrosis, most probably due to CLP36 accumulation in cardiomyocytes. Altogether, this work reveals autophagy may protect cardiomyocytes from the MI/R injury through the clearance of CLP36, and these findings define a novel relationship between autophagy and the regulation of stress fibre in heart.
Mucopolysaccharidosis type IIIC (MPSIIIC) is a severe lysosomal storage disease caused by deficiency in activity of the transmembrane enzyme heparan-a-glucosaminide N-acetyltransferase (HGSNAT) that catalyses the N-acetylation of a-glucosamine residues of heparan sulfate. Enzyme deficiency causes abnormal substrate accumulation in lysosomes, leading to progressive and severe neurodegeneration, somatic pathology and early death. There is no cure for MPSIIIC, and development of new therapies is challenging because of the unfeasibility of cross-correction. In this study, we generated a new mouse model of MPSIIIC by targeted disruption of the Hgsnat gene. Successful targeting left LacZ expression under control of the Hgsnat promoter, allowing investigation into sites of endogenous expression, which was particularly prominent in the CNS, but was also detectable in peripheral organs. Signs of CNS storage pathology, including glycosaminoglycan accumulation, lysosomal distension, lysosomal dysfunction and neuroinflammation were detected in 2-month-old animals and progressed with age. Glycosaminoglycan accumulation and ultrastructural changes were also observed in most somatic organs, but lysosomal pathology seemed most severe in liver. Furthermore, HGSNAT-deficient mice had altered locomotor and exploratory activity and shortened lifespan. Hence, this animal model recapitulates human MPSIIIC and provides a useful tool for the study of disease physiopathology and the development of new therapeutic approaches.
An early event in Alzheimer's disease (AD) pathogenesis is the formation of extracellular aggregates of amyloid-ß peptide (Aß), thought to be initiated by a prion-like seeding mechanism. However, the molecular nature and location of the Aß seeds remain rather elusive. Active Aß seeds are found in crude homogenates of amyloid-laden brains and in the soluble fraction thereof. To analyze the seeding activity of the pellet fraction, we have either separated or directly immunoisolated membranes from such homogenates. Here, we found considerable Aß seeding activity associated with membranes in the absence of detectable amyloid fibrils. We also found that Aß seeds on mitochondrial or associated membranes efficiently induced Aß aggregation in vitro and seed ß-amyloidosis in vivo. Aß seeds at intracellular membranes may contribute to the spreading of Aß aggregation along neuronal pathways and to the induction of intracellular pathologies downstream of Aß.
Acupuncture has historically been practiced to treat medical disorders by mechanically stimulating specific acupoints with fine needles. Despite its well-documented efficacy, its biological basis remains largely elusive. In this study, we found that mechanical stimulation at the acupoint of Yanglingquan (GB34) promoted the autophagic clearance of a-synuclein (a-syn), a well known aggregation-prone protein closely related to Parkinson's disease (PD), in the substantia nigra par compacta (SNpc) of the brain in a PD mouse model. We found the protein clearance arose from the activation of the autophagy-lysosome pathway (ALP) in a mammalian target of rapamycin (mTOR)-independent approach. Further, we observed the recovery in the activity of dopaminergic neurons in SNpc, and improvement in the motor function at the behavior level of PD mice. Whereas acupuncture and rapamycin, a chemical mTOR inhibitor, show comparable a-syn clearance and therapeutic effects in the PD mouse model, the latter adopts a distinctly different, mTOR-dependent, autophagy induction process. Due to this fundamental difference, acupuncture may circumvent adverse effects of the rapamycin treatment. The newly discovered connection between acupuncture and autophagy not only provides a new route to understanding the molecular mechanism of acupuncture but also sheds new light on cost-effective and safe therapy of neurodegenerative diseases.
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease worldwide. Gpnmb is classified as a type 1 membrane protein and its soluble form is secreted by ADAM10-mediated cleavage. Gpnmb mRNA was found in the Kupffer cells and white adipose tissues (WATs) and its upregulation in obesity was recently found. Here, we generated aP2 promoter-driven Gpnmb transgenic (Tg) mice and the overexpression of Gpnmb ameliorated the fat accumulation and fibrosis of the liver in diet-induced obesity model. Soluble form of Gpnmb in sera was elevated in Gpnmb Tg mice and Gpnmb concentrated in hepatic macrophages and stellate cells interacted with calnexin, which resulted in the reduction of oxidative stress. In the patients with non-alcoholic steatohepatitis, serum soluble GPNMB concentrations were higher compared with the patients with simple steatosis. The GPNMB is a promising biomarker and therapeutic target for the development and progression of NAFLD in obesity.
Mammalian cells possess two amino acid-sensing kinases: general control nonderepressible 2 (GCN2) and mechanistic target of rapamycin complex 1 (mTORC1). Their combined effects orchestrate cellular adaptation to amino acid levels, but how their activities are coordinated remains poorly understood. Here, we demonstrate an important link between GCN2 and mTORC1 signaling. Upon deprivation of various amino acids, activated GCN2 up-regulates ATF4 to induce expression of the stress response protein Sestrin2, which is required to sustain repression of mTORC1 by blocking its lysosomal localization. Moreover, Sestrin2 induction is necessary for cell survival during glutamine deprivation, indicating that Sestrin2 is a critical effector of GCN2 signaling that regulates amino acid homeostasis through mTORC1 suppression.
Endosomes, lysosomes and related catabolic organelles are a dynamic continuum of vacuolar structures that impact a number of cell physiological processes such as protein/lipid metabolism, nutrient sensing and cell survival. Here we develop a library of ultra-pH-sensitive fluorescent nanoparticles with chemical properties that allow fine-scale, multiplexed, spatio-temporal perturbation and quantification of catabolic organelle maturation at single organelle resolution to support quantitative investigation of these processes in living cells. Deployment in cells allows quantification of the proton accumulation rate in endosomes; illumination of previously unrecognized regulatory mechanisms coupling pH transitions to endosomal coat protein exchange; discovery of distinct pH thresholds required for mTORC1 activation by free amino acids versus proteins; broad-scale characterization of the consequence of endosomal pH transitions on cellular metabolomic profiles; and functionalization of a context-specific metabolic vulnerability in lung cancer cells. Together, these biological applications indicate the robustness and adaptability of this nanotechnology-enabled 'detection and perturbation' strategy.
Cyclosporine A (CsA) is widely used as an immunosuppressor in transplantation. Previous studies reported that CsA induces autophagy and that chronic treatment with CsA results in accumulation of autophagosomes and reduced autophagic clearance. Autophagy is a prosurvival process that promotes recovery from acute kidney injury by degrading misfolded proteins produced in the kidney. In the present study, we used TMBIM6-expressing HK-2, human kidney tubular cells (TMBIM6 cells) and Tmbim6 knockout (tmbim6(-/-)) mice. When exposed to CsA, the TMBIM6 cells maintained autophagy activity by preventing autophagosome accumulation. With regard to signaling, PRKKA/AMPK phosphorylation and mechanistic target of rapamycin (serine/threonine kinase) complex 1 (MTORC1) expression and its downstream target TFEB (transcription factor EB), a lysosome biogenesis factor, were regulated in the TMBIM6 cells. Lysosomal activity was highly increased or stably maintained in the presence of TMBIM6. In addition, treatment of tmbim6(-/-) mice with CsA resulted in increased autophagosome formation and decreased lysosome formation and activity. We also found that tmbim6(-/-) mice were susceptible to CsA-induced kidney injury. Taken together, these results indicate that TMBIM6 protects against CsA-induced nephrotoxicity both in vitro and in vivo by inducing autophagy and activating lysosomes.
Myocyte function and survival relies on the maintenance of a healthy population of mitochondria. The PINK1/Parkin pathway plays an important role in clearing defective mitochondria via autophagy in cells. However, how the PINK1/Parkin pathway regulates mitochondrial quality control and whether it coordinates with other mitophagy pathways are still unclear. Therefore, the objective of this study was to investigate the effect of PINK1-deficiency on mitochondrial quality control in myocytes. Using PINK1-deficient (PINK1-/-) mice, we found that Parkin is recruited to damaged cardiac mitochondria in hearts after treatment with the mitochondrial uncoupler FCCP or after a myocardial infarction even in the absence of PINK1. Parkin recruitment to depolarized mitochondria correlates with increased ubiquitination of mitochondrial proteins and activation of mitophagy in PINK1-/- myocytes. In addition, induction of mitophagy by the atypical BH3-only protein BNIP3 is unaffected by lack of PINK1. Overall, these data suggest that Parkin recruitment to depolarized cardiac mitochondria and subsequent activation of mitophagy is independent of PINK1. Moreover, alternative mechanisms of Parkin activation and pathways of mitophagy remain functional in PINK1-/- myocytes and could compensate for the PINK1 deficiency.
High glucose reduces autophagy and enhances apoptosis of podocytes. Previously, we reported that high glucose induced podocyte injury through upregulation of the (pro)renin receptor (PRR). We hypothesized that increasing PRR reduces autophagy and increases apoptosis of mouse podocytes exposed to high glucose via activation of the PI3K/Akt/mTOR signaling pathway. Mouse podocytes were cultured in normal (5 mmol/l) or high (25 mmol/l) d-glucose for 48 h. High glucose significantly increased mRNA and protein levels of PRR, phosphorylation of PI3K/Akt/mTOR, and p62. In contrast, high glucose decreased activation of UNC-51-like kinase-1 (ULK1) by phosphorylating Ser757 and protein levels of microtubule-associated protein-1 light chain 3B (LC3B)-II and Lamp-2. Bafilomycin A1 increased LC3BII and p62 accumulation in high-glucose-treated cells. High glucose reduced the autophagic flux. Confocal microscopy studies showed significant reduction in the protein level of LC3B in response to high glucose. Cyto-ID autophagy staining showed a significant decrease in autophagosome formation with high glucose. In the absence of PRR, activation of Akt with sc-79 or mTOR with MHY-1485 increased p62 accumulation. Caspase-3/7 activity and apoptosis monitored by TUNEL assay were significantly increased in podocytes treated with high glucose. PRR siRNA significantly reversed the effects of high glucose. Based on these data, we conclude that high glucose decreases autophagy and increases apoptosis in mouse podocytes through the PRR/PI3K/Akt/mTOR signaling pathway.
High-fat diet (HFD) and inflammation are the key contributors to insulin resistance and type 2 diabetes (T2D). Previous study shows fatty acid-induced accumulation of damaged, reactive oxygen species (ROS)-generating mitochondria, and this in turn activates the NLRP3 inflammasome interference with insulin signaling. Our previous research shows NLRP3 inflammasome activation signal originates from defects in autophagy. Yet how the fatty acid related to mitophagy alteration leads to the activation of NLRP3-ASC inflammasome has not been considered. Here we demonstrated that palmitate (PA) induced mitophagy deficiency, leading to damaged mitochondrion as characterized by mito-ROS production and loss of membrane potential. Antioxidant APDC or Ca(2+) signaling inhibitor Nifedipine blocked PA-induced NLRP3 inflammasome activation. Further, we provided evidences that PA reduced the expression of Ras homolog enriched in brain (Rheb) and disrupted Rheb recruitment to the mitochondrial outer membrane. In addition, sustained PA caused disassociation of kinesin family member 5B (KIF5B) from binding with mitochondria via Ca(2+)-dependent effects. Disruption of Rheb and KIF5B interaction with mitochondria blocked mitochondrial degradation along with IL-1ß dependent insulin resistance, which was majorly attenuated by Rheb/KIF5B overexpression. In a consequence, defective mitophagy led to the accumulation of damaged-ROS-generating mitochondria, down pathway of NLRP3-ASC-Caspase 1 activation, and subsequently, insulin resistance. These findings provide insights into the association of inflammation, mitophagy and T2D.
We have previously demonstrated that Stat3 regulates lysosomal-mediated programmed cell death (LM-PCD) during mouse mammary gland involution in vivo. However, the mechanism that controls the release of lysosomal cathepsins to initiate cell death in this context has not been elucidated. We show here that Stat3 regulates the formation of large lysosomal vacuoles that contain triglyceride. Furthermore, we demonstrate that milk fat globules (MFGs) are toxic to epithelial cells and that, when applied to purified lysosomes, the MFG hydrolysate oleic acid potently induces lysosomal leakiness. Additionally, uptake of secreted MFGs coated in butyrophilin 1A1 is diminished in Stat3-ablated mammary glands and loss of the phagocytosis bridging molecule MFG-E8 results in reduced leakage of cathepsins in vivo. We propose that Stat3 regulates LM-PCD in mouse mammary gland by switching cellular function from secretion to uptake of MFGs. Thereafter, perturbation of lysosomal vesicle membranes by high levels of free fatty acids results in controlled leakage of cathepsins culminating in cell death.
Myoblast fusion (a critical process by which muscles grow) occurs in a multi-step fashion that requires actin and membrane remodeling; but important questions remain regarding the spatial/temporal regulation of and interrelationship between these processes. We recently reported that the Rho-GAP, GRAF1, was particularly abundant in muscles undergoing fusion to form multinucleated fibers and that enforced expression of GRAF1 in cultured myoblasts induced robust fusion by a process that required GAP-dependent actin remodeling and BAR domain-dependent membrane sculpting. Herein we developed a novel line of GRAF1-deficient mice to explore a role for this protein in the formation/maturation of myotubes in vivo. Post-natal muscles from GRAF1-depleted mice exhibited a significant and persistent reduction in cross-sectional area, impaired regenerative capacity and a significant decrease in force production indicative of lack of efficient myoblast fusion. A significant fusion defect was recapitulated in isolated myoblasts depleted of GRAF1 or its closely related family member GRAF2. Mechanistically, we show that GRAF1 and 2 facilitate myoblast fusion, at least in part, by promoting vesicle-mediated translocation of fusogenic ferlin proteins to the plasma membrane.
Copyright © 2014. Published by Elsevier Inc.
The role of remote astrocyte (AC) reaction to central or peripheral axonal insult is not clearly understood. Here we use a transgenic approach to compare the direct influence of normal with diminished AC reactivity on neuronal integrity and synapse recovery following extracranial facial nerve transection in mice. Our model allows straightforward interpretations of AC-neuron signalling by reducing confounding effects imposed by inflammatory cells. We show direct evidence that perineuronal reactive ACs play a major role in maintaining neuronal circuitry following distant axotomy. We reveal a novel function of astrocytic signal transducer and activator of transcription-3 (STAT3). STAT3 regulates perineuronal astrocytic process formation and re-expression of a synaptogenic molecule, thrombospondin-1 (TSP-1), apart from supporting neuronal integrity. We demonstrate that, through this new pathway, TSP-1 is responsible for the remote AC-mediated recovery of excitatory synapses onto axotomized motor neurons in adult mice. These data provide new targets for neuroprotective therapies via optimizing AC-driven plasticity.
In the latter half of the 20th century, societal and technological changes led to a shift in the composition of the American diet to include a greater proportion of processed, pre-packaged foods high in fat and carbohydrates, and low in dietary fiber (a "Western diet"). Over the same time period, there have been parallel increases in Salmonella gastroenteritis cases and a broad range of chronic inflammatory diseases associated with intestinal dysbiosis. Several polysaccharide food additives are linked to bacterially-driven intestinal inflammation and may contribute to the pathogenic effects of a Western diet. Therefore, we examined the effect of a ubiquitous polysaccharide food additive, maltodextrin (MDX), on clearance of the enteric pathogen Salmonella using both in vitro and in vivo infection models. When examined in vitro, murine bone marrow-derived macrophages exposed to MDX had altered vesicular trafficking, suppressed NAPDH oxidase expression, and reduced recruitment of NADPH oxidase to Salmonella-containing vesicles, which resulted in persistence of Salmonella in enlarged Rab7+ late endosomal vesicles. In vivo, mice consuming MDX-supplemented water had a breakdown of the anti-microbial mucous layer separating gut bacteria from the intestinal epithelium surface. Additionally, oral infection of these mice with Salmonella resulted in increased cecal bacterial loads and enrichment of lamina propria cells harboring large Rab7+ vesicles. These findings indicate that consumption of processed foods containing the polysaccharide MDX contributes to suppression of intestinal anti-microbial defense mechanisms and may be an environmental priming factor for the development of chronic inflammatory disease.
Diabetic neuropathy develops on a background of hyperglycemia and an entangled metabolic imbalance. There is increasing evidence of central nervous system involvement in diabetic neuropathy and no satisfactory treatment except maintenance of good glycemic control, thereby highlighting the importance of identifying novel therapeutic targets. Purkinje cells are a class of metabolically specialized active neurons, and degeneration of Purkinje cells is a common feature of inherited ataxias in humans and mice. However, whether Purkinje cells are implicated in diabetic neuropathy development under metabolic stress remains poorly defined. Here, we revealed a novel leucine-rich repeat kinase 2 (LRRK2)-mediated pathway in Purkinje cells that is involved in the pathogenesis of diabetic neuropathy from a 24-week long study of streptozotocin (STZ)-diabetic rats. We found that hyperglycemia, cerebellum proinflammatory cytokines, and chemokines increased markedly in 24-week STZ-diabetic rats. Furthermore, we demonstrated that degeneration of Purkinje cells is characterized by progressive swellings of axon terminals, no autophagosome formation, the reduction of LC3II/LC3I and Lamp2, and accumulation of p62 puncta in 24-week STZ-diabetic rats. Importantly, a higher expression level of LRRK2-mediated hyperphosphorylation of tau along with increased mitochondrial dynamin-like protein (mito-DLP1) was demonstrated in 24-week STZ-diabetic rats. This effect of LRRK2 overexpression induced mitochondrial fragmentation, and reduced mitochondrial protein degradation rates were confirmed in vitro. As a consequence, 24-week STZ-diabetic rats showed mitochondrial dysfunction in cerebellar Purkinje neurons and coordinated motor deficits evaluated by rotarod test. Our findings are to our knowledge the first to suggest that the LRRK2-mediated pathway induces mitochondrial dysfunction and loss of cerebellar Purkinje neurons and, subsequently, may be associated with motor coordination deficits in STZ-diabetic rats. These data may indicate a novel cellular therapeutic target for diabetic neuropathy.
EHD proteins have been implicated in intracellular trafficking, especially endocytic recycling, where they mediate receptor and lipid recycling back to the plasma membrane. Additionally, EHDs help regulate cytoskeletal reorganization and induce tubule formation. It was previously shown that EHD proteins bind directly to the C2 domains in myoferlin, a protein that regulates myoblast fusion. Loss of myoferlin impairs normal myoblast fusion leading to smaller muscles in vivo but the intracellular pathways perturbed by loss of myoferlin function are not well known. We now characterized muscle development in EHD1-null mice. EHD1-null myoblasts display defective receptor recycling and mislocalization of key muscle proteins, including caveolin-3 and Fer1L5, a related ferlin protein homologous to myoferlin. Additionally, EHD1-null myoblast fusion is reduced. We found that loss of EHD1 leads to smaller muscles and myofibers in vivo. In wildtype skeletal muscle EHD1 localizes to the transverse tubule (T-tubule), and loss of EHD1 results in overgrowth of T-tubules with excess vesicle accumulation in skeletal muscle. We provide evidence that tubule formation in myoblasts relies on a functional EHD1 ATPase domain. Moreover, we extended our studies to show EHD1 regulates BIN1 induced tubule formation. These data, taken together and with the known interaction between EHD and ferlin proteins, suggests that the EHD proteins coordinate growth and development likely through mediating vesicle recycling and the ability to reorganize the cytoskeleton.
Degradation of damaged mitochondria by mitophagy is an essential process to ensure cell homeostasis. Because neurons, which have a high energy demand, are particularly dependent on the mitochondrial dynamics, mitophagy represents a key mechanism to ensure correct neuronal function. Collapsin response mediator proteins 5 (CRMP5) belongs to a family of cytosolic proteins involved in axon guidance and neurite outgrowth signaling during neural development. CRMP5, which is highly expressed during brain development, plays an important role in the regulation of neuronal polarity by inhibiting dendrite outgrowth at early developmental stages. Here, we demonstrated that CRMP5 was present in vivo in brain mitochondria and is targeted to the inner mitochondrial membrane. The mitochondrial localization of CRMP5 induced mitophagy. CRMP5 overexpression triggered a drastic change in mitochondrial morphology, increased the number of lysosomes and double membrane vesicles termed autophagosomes, and enhanced the occurrence of microtubule-associated protein 1 light chain 3 (LC3) at the mitochondrial level. Moreover, the lipidated form of LC3, LC3-II, which triggers autophagy by insertion into autophagosomes, enhanced mitophagy initiation. Lysosomal marker translocates at the mitochondrial level, suggesting autophagosome-lysosome fusion, and induced the reduction of mitochondrial content via lysosomal degradation. We show that during early developmental stages the strong expression of endogenous CRMP5, which inhibits dendrite growth, correlated with a decrease of mitochondrial content. In contrast, the knockdown or a decrease of CRMP5 expression at later stages enhanced mitochondrion numbers in cultured neurons, suggesting that CRMP5 modulated these numbers. Our study elucidates a novel regulatory mechanism that utilizes CRMP5-induced mitophagy to orchestrate proper dendrite outgrowth and neuronal function.
The cytokines RANKL and TNF activate NF-?B signaling in osteoclast precursors (OCPs) to induce osteoclast (OC) formation. Conversely, TNF can limit OC formation through NF-?B p100, which acts as an inhibitor, and TNF receptor-associated receptor 3 (TRAF3); however, a role for TRAF3 in RANKL-mediated OC formation is unknown. We found that TRAF3 limits RANKL-induced osteoclastogenesis by suppressing canonical and noncanonical NF-?B signaling. Conditional OC-specific Traf3-KO (cKO) mice had mild osteoporosis and increased OC formation. RANKL induced TRAF3 degradation via the lysosome/autophagy system. The autophagy/lysosome inhibitor chloroquine reduced RANKL-induced OC formation and function by increasing TRAF3 expression in OCPs in vitro and in vivo. Although chloroquine had no effect on basal bone resorption, it inhibited parathyroid hormone- and ovariectomy-induced OC activation in WT, but not cKO, mice. Deletion of the transcription factor gene Relb resulted in increased TRAF3 expression in OCPs, which was associated with decreased RANKL-induced TRAF3 degradation. RelB directly increased expression of BECN1, a key autophagy regulator, by binding to its promoter. These data indicate that autophagic/lysosomal degradation of TRAF3 is an important step in RANKL-induced NF-?B activation in OCPs. Furthermore, treatments that increase TRAF3 levels in OCPs, including pharmacological inhibition of its degradation with compounds such as chloroquine, may limit bone destruction in common bone diseases.
An acute bout of exercise activates downstream signaling cascades that ultimately result in mitochondrial biogenesis. In addition to inducing mitochondrial synthesis, exercise triggers the removal of damaged cellular material via autophagy and of dysfunctional mitochondria through mitophagy. Here, we investigated the necessity of p53 to the changes that transpire within the muscle upon an imposed metabolic and physiological challenge, such as a bout of endurance exercise. We randomly assigned wild-type (WT) and p53 knockout (KO) mice to control, acute exercise (AE; 90 min at 15 m/min), and AE + 3 h recovery (AER) groups and measured downstream alterations in markers of mitochondrial biogenesis, autophagy, and mitophagy. In the absence of p53, activation of p38 MAPK upon exercise was abolished, whereas CaMKII and AMP-activated protein kinase only displayed an attenuated enhancement in the AER group compared with WT mice. The translocation of peroxisome proliferator-activated receptor-? coactivator-1 a to the nucleus was diminished and only observed in the AER group, and the subsequent increase in messenger RNA transcripts related to mitochondrial biogenesis with exercise and recovery was absent in the p53 KO animals. Whole-muscle autophagic and lysosomal markers did not respond to exercise, irrespective of the genotype of the exercised mice, with the exception of increased ubiquitination observed in KO mice with exercise. Markers of mitophagy were elevated in response to AE and AER conditions in both WT and p53 KO runners. The data suggest that p53 is important for the exercise-induced activation of mitochondrial synthesis and is integral in regulating autophagy during control conditions but not in response to exercise.
Impairment of glucose-stimulated insulin secretion caused by the lipotoxicity of palmitate was found in ß-cells. Recent studies have indicated that defects in autophagy contribute to pathogenesis in type 2 diabetes. Here, we report that autophagy-related 7 (Atg7) induced excessive autophagic activation in INS-1(823/13) cells exposed to saturated fatty acids. Atg7-induced cathepsin B (CTSB) overexpression resulted in an unexpected significant increase in proinflammatory chemokine and cytokine production levels of IL-1ß, monocyte chemotactic protein-1, IL-6, and TNF-a. Inhibition of receptor-interacting protein did not affect the inflammatory response, ruling out involvement of necrosis. CTSB siRNA suppressed the inflammatory response but did not affect apoptosis significantly, suggesting that CTSB was a molecular linker between autophagy and the proinflammatory response. Blocking caspase-3 suppressed apoptosis but did not affect the inflammatory response, suggesting that CTSB induced inflammatory effects independently of apoptosis. Silencing of Nod-like receptor 3 (NLRP3) completely abolished both IL-1ß secretion and the down-regulation effects of Atg7-induced CTSB overexpression on glucose-stimulated insulin secretion impairment, thus identifying the NLRP3 inflammasome as an autophagy-responsive element in the pancreatic INS-1(823/13) cell line. Combined together, our results indicate that CTSB contributed to the Atg7-induced NLRP3-dependent proinflammatory response, resulting in aggravation of lipotoxicity, independently of apoptosis in the pancreatic INS-1(823/13) cell line.
Mucopolysaccharidosis (MPS) are a family of related disorders caused by a mutation in one of the lysosomal exoglycosidases which leads to the accumulation of glycosaminoglycans (GAGs). MPS VII, caused by a mutation in ß-glucuronidase, manifests hepatomegaly, skeletal dysplasia, short stature, corneal clouding, and developmental delay. Current treatment regimens for MPS are not effective for treating corneal clouding and impaired mental development. We hypothesized that human umbilical mesenchymal stem cells (UMSCs) transplanted into the corneal stroma could participate in the catabolism of GAGs providing a means of cell therapy for MPS. For such treatment, human UMSCs were intrastromally transplanted into corneas of MPS VII mice. UMSC transplantation restored the dendritic and hexagonal morphology of host keratocytes and endothelial cells, respectively, and in vivo confocal microscopy (HRT-II) revealed reduced corneal haze. Immunohistochemistry using antibodies against heparan sulfate and chondroitin sulfate chains as well as lysosomal-associated membrane protein 2 revealed a decrease in GAG content and both lysosomal number and size in the treated corneas. Labeling UMSC intracellular compartments prior to transplantation revealed the distribution of UMSC vesicles throughout the corneal stroma and endothelium. An in vitro coculture assay between skin fibroblasts isolated from MPS VII mice and UMSC demonstrated that neutral vesicles released by the UMSC are taken up by the fibroblasts and proceed to fuse with the acidic lysosomes. Therefore, transplanted UMSCs participate both in extracellular GAG turnover and enable host keratocytes to catabolize accumulated GAG products, suggesting that UMSC could be a novel alternative for treating corneal defects associated with MPS and other congenital metabolic disorders.
The goal of this study was to determine whether the mechanical activation of mechanistic target of rapamycin (mTOR) signalling is associated with changes in phosphorylation of tuberous sclerosis complex-2 (TSC2) and targeting of mTOR and TSC2 to the lysosome. As a source of mechanical stimulation, mouse skeletal muscles were subjected to eccentric contractions (ECs). The results demonstrated that ECs induced hyper-phosphorylation of TSC2 and at least part of this increase occurred on residue(s) that fall within RxRxxS/T consensus motif(s). Furthermore, in control muscles, we found that both mTOR and TSC2 are highly enriched at the lysosome. Intriguingly, ECs enhanced the lysosomal association of mTOR and almost completely abolished the lysosomal association of TSC2. Based on these results, we developed a new model that could potentially explain how mechanical stimuli activate mTOR signalling. Furthermore, this is the first study to reveal that the activation of mTOR is associated with the translocation of TSC2 away from the lysosome. Since a large number of signalling pathways rely on TSC2 to control mTOR signalling, our results have potentially revealed a fundamental mechanism via which not only mechanical, but also various other types of stimuli, control mTOR signalling.
Mammalian Target of Rapamycin Complex 1 (mTORC1) is activated by growth factor-regulated phosphoinositide 3-kinase (PI3K)/Akt/Rheb signalling and extracellular amino acids (AAs) to promote growth and proliferation. These AAs induce translocation of mTOR to late endosomes and lysosomes (LELs), subsequent activation via mechanisms involving the presence of intralumenal AAs, and interaction between mTORC1 and a multiprotein assembly containing Rag GTPases and the heterotrimeric Ragulator complex. However, the mechanisms by which AAs control these different aspects of mTORC1 activation are not well understood. We have recently shown that intracellular Proton-assisted Amino acid Transporter 1 (PAT1)/SLC36A1 is an essential mediator of AA-dependent mTORC1 activation. Here we demonstrate in Human Embryonic Kidney (HEK-293) cells that PAT1 is primarily located on LELs, physically interacts with the Rag GTPases and is required for normal AA-dependent mTOR relocalisation. We also use the powerful in vivo genetic methodologies available in Drosophila to investigate the regulation of the PAT1/Rag/Ragulator complex. We show that GFP-tagged PATs reside at both the cell surface and LELs in vivo, mirroring PAT1 distribution in several normal mammalian cell types. Elevated PI3K/Akt/Rheb signalling increases intracellular levels of PATs and synergistically enhances PAT-induced growth via a mechanism requiring endocytosis. In light of the recent identification of the vacuolar H(+)-ATPase as another Rag-interacting component, we propose a model in which PATs function as part of an AA-sensing engine that drives mTORC1 activation from LEL compartments.
Our aim was to elucidate the physiological role of calpains (CAPN) in mammary gland involution. Both CAPN-1 and -2 were induced after weaning and its activity increased in isolated mitochondria and lysosomes. CAPN activation within the mitochondria could trigger the release of cytochrome c and other pro-apoptotic factors, whereas in lysosomes it might be essential for tissue remodeling by releasing cathepsins into the cytosol. Immunohistochemical analysis localized CAPNs mainly at the luminal side of alveoli. During weaning, CAPNs translocate to the lysosomes processing membrane proteins. To identify these substrates, lysosomal fractions were treated with recombinant CAPN and cleaved products were identified by 2D-DIGE. The subunit b(2) of the v-type H(+) ATPase is proteolyzed and so is the lysosomal-associated membrane protein 2a (LAMP2a). Both proteins are also cleaved in vivo. Furthermore, LAMP2a cleavage was confirmed in vitro by addition of CAPNs to isolated lysosomes and several CAPN inhibitors prevented it. Finally, in vivo inhibition of CAPN1 in 72-h-weaned mice decreased LAMP2a cleavage. Indeed, calpeptin-treated mice showed a substantial delay in tissue remodeling and involution of the mammary gland. These results suggest that CAPNs are responsible for mitochondrial and lysosomal membrane permeabilization, supporting the idea that lysosomal-mediated cell death is a new hallmark of mammary gland involution.
Loss-of-function mutations in dysferlin cause muscular dystrophy, and dysferlin has been implicated in resealing membrane disruption in myofibers. Given the importance of membrane fusion in many aspects of muscle function, we studied the role of dysferlin in muscle growth. We found that dysferlin null myoblasts have a defect in myoblast-myotube fusion, resulting in smaller myotubes in culture. In vivo, dysferlin null muscle was found to have mislocalized nuclei and vacuolation. We found that myoblasts isolated from dysferlin null mice accumulate enlarged, lysosomal-associated membrane protein 2 (LAMP2)-positive lysosomes. Dysferlin null myoblasts accumulate transferrin-488, reflecting abnormal vesicular trafficking. Additionally, dysferlin null myoblasts display abnormal trafficking of the insulin-like growth factor (IGF) receptor, where the receptor is shuttled to LAMP2-positive lysosomes. We studied growth, in vivo, by infusing mice with the growth stimulant IGF1. Control IGF1-treated mice increased myofiber diameter by 30% as expected, whereas dysferlin null muscles had no response to IGF1, indicating a defect in myofiber growth. We also noted that dysferlin null fibroblasts also accumulate acidic vesicles, IGF receptor and transferrin, indicating that dysferlin is important for nonmuscle vesicular trafficking. These data implicate dysferlin in multiple membrane fusion events within the cell and suggest multiple pathways by which loss of dysferlin contributes to muscle disease.
Insulin-like growth factor (IGF) is a potent stimulus of muscle growth. Myoferlin is a membrane-associated protein important for muscle development and regeneration. Myoferlin-null mice have smaller muscles and defective myoblast fusion. To understand the mechanism by which myoferlin loss retards muscle growth, we found that myoferlin-null muscle does not respond to IGF1. In vivo after IGF1 infusion, control muscle increased myofiber diameter by 25%, but myoferlin-null muscle was unresponsive. Myoblasts cultured from myoferlin-null muscle and treated with IGF1 also failed to show the expected increase in fusion to multinucleate myotubes. The IGF1 receptor colocalized with myoferlin at sites of myoblast fusion. The lack of IGF1 responsiveness in myoferlin-null myoblasts was linked directly to IGF1 receptor mistrafficking as well as decreased IGF1 signaling. In myoferlin-null myoblasts, the IGF1 receptor accumulated into large vesicular structures. These vesicles colocalized with a marker of late endosomes/lysosomes, LAMP2, specifying redirection from a recycling to a degradative pathway. Furthermore, ultrastructural analysis showed a marked increase in vacuoles in myoferlin-null muscle. These data demonstrate that IGF1 receptor recycling is required for normal myogenesis and that myoferlin is a critical mediator of postnatal muscle growth mediated by IGF1.-Demonbreun, A. R., Posey, A. D., Heretis, K., Swaggart, K. A., Earley, J. U., Pytel, P., McNally, E. M. Myoferlin is required for insulin-like growth factor response and muscle growth.
Functional atrophy and accompanying lymphocytic infiltration and destruction of the lacrimal gland (LG) are characteristics of Sjögren's Syndrome (SjS). The male NOD mouse is an experimental model for the autoimmune exocrinopathy that develops in the LG of SjS patients. Acinar cells in LG of male NOD mice aged 3-4 months were previously shown to accumulate lipid droplets. In the current study, analysis of lipid components revealed that the accumulated lipids were mostly cholesteryl esters (CE). Gene expression microarray analysis followed by real-time RT-PCR revealed alterations in the expression of several genes involved in lipid homeostasis in LG of 12-week-old male NOD mice relative to matched BALB/c controls. A series of upregulated genes including apolipoprotein E, apolipoprotein F, hepatic lipase, phosphomevalonate kinase, ATP-binding cassette D1 and ATP-binding cassette G1 were identified. Comparison of liver mRNAs to LG mRNAs in BALB/c and NOD mice revealed that the differential expressions were LG-specific. Gene expression profiles were also characterized in LGs of female mice, younger mice and immune-incompetent NOD SCID mice. Investigation of the cellular distribution of Apo-E and Apo-F proteins suggested that these proteins normally coordinate to mediate lipid efflux from the acinar cells but that dysfunction of these processes due to missorting of Apo-F may contribute to CE deposition. Finally, the initiation and extent of lipid deposition were correlated with lymphocytic infiltration in the LG of male NOD mice. We propose that impaired lipid efflux contributes to lipid deposition, an event that may contribute to the development and/or progression of dacryoadenitis in the male NOD mouse.
The mechanisms through which the small GTPases Rac1 and Cdc42 regulate the formation of membrane ruffles, lamellipodia, and filopodia are currently unknown. The p21-activated kinases (PAKs) are direct targets of active Rac and Cdc42 which can induce the assembly of polarized cytoskeletal structures when expressed in fibroblasts, suggesting that they may play a role in mediating the effects of these GTPases on cytoskeletal dynamics. We have examined the subcellular localization of endogenous PAK1 in fibroblast cell lines using specific PAK1 antibodies. PAK1 is detected in submembranous vesicles in both unstimulated and stimulated fibroblasts that colocalize with a marker for fluid-phase uptake. In cells stimulated with PDGF, in v-Src-transformed fibroblasts, and in wounded cells, PAK1 redistributed into dorsal and membrane ruffles and into the edges of lamellipodia, where it colocalizes with polymerized actin. PAK1 was also colocalized with F-actin in membrane ruffles extended as a response to constitutive activation of Rac1. PAK1 appears to precede F-actin in translocating to cytoskeletal structures formed at the cell periphery. The association of PAK1 with the actin cytoskeleton is prevented by the actin filament-disrupting agent cytochalasin D and by the phosphatidylinositol 3-kinase inhibitor wortmannin. Co-immunoprecipitation experiments demonstrate an in vivo interaction of PAK1 with filamentous (F)-actin in stimulated cells. Microinjection of a constitutively active PAK1 mutant into Rat-1 fibroblasts overexpressing the insulin receptor (HIRcB cells) induced the formation of F-actin- and PAK1-containing structures reminiscent of dorsal ruffles. These data indicate a close correlation between the subcellular distribution of endogenous PAK1 and the formation of Rac/Cdc42-dependent cytoskeletal structures and support an active role for PAK1 in regulating cortical actin rearrangements.
Purpose: Lacrimal glands (LGs) of male NOD mice, a model of Sjögren's syndrome (SjS), exhibit immune cell infiltration and lipid deposition. The mechanism of SjS was further investigated by characterizing gene expression profiles of NOD mouse LGs in comparison with those of healthy control mice. Differentially expressed genes were further investigated at the protein level to correlate changes in location and abundance with development of disease.
Methods: Microarray followed by real-time RT-PCR was conducted to compare the gene expression in 12-week-old male NOD mouse LG relative to that in matched BALB/c mouse LG. Immunofluorescence and Western blot analyses were used to localize and quantify proteins of interest. Enzymatic assays measured catalytic activity of cathepsins.
Results: Cathepsin H (Ctsh), S (Ctss), and Z (Ctsz) and proinflammatory factors, including tumor necrosis factor (Tnf), interleukin 6 (Il6), and interleukin 1 beta (Il1b), were upregulated at the mRNA level. Increased cathepsin S immunofluorescence was detected in lysosomes and secretory vesicle-like organelles in LG acinar cells and CD68-positive infiltrating macrophages in NOD mouse LG. Cathepsin S (CATS) and cathepsin H (CATH) activities were significantly higher in NOD mouse LG lysate than in control lysates, and CATS was also significantly elevated in NOD mouse tears.
Conclusions: Expression of CATS and CATH increases in parallel with proinflammatory cytokines during the development of autoimmune inflammatory disease in the NOD mouse disease model. Tear CATS may represent a biomarker for diagnosis of dacryoadenitis in SjS.
BACKGROUND:
In multiple sclerosis, inflammation can successfully be prevented, while promoting repair is still a major challenge. Microglial cells, the resident phagocytes of the central nervous system (CNS), are hematopoietic-derived myeloid cells and express the triggering receptor expressed on myeloid cells 2 (TREM2), an innate immune receptor. Myeloid cells are an accessible source for ex vivo gene therapy. We investigated whether myeloid precursor cells genetically modified to express TREM2 affect the disease course of experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis.
METHODS AND FINDINGS:
EAE was induced in mice by immunization with a myelin autoantigen. Intravenous application of TREM2-transduced bone marrow-derived myeloid precursor cells at the EAE peak led to an amelioration of clinical symptoms, reduction in axonal damage, and prevention of further demyelination. TREM2-transduced myeloid cells applied intravenously migrated into the inflammatory spinal cord lesions of EAE-diseased mice, showed increased lysosomal and phagocytic activity, cleared degenerated myelin, and created an anti-inflammatory cytokine milieu within the CNS.
CONCLUSIONS:
Intravenously applied bone marrow-derived and TREM2-tranduced myeloid precursor cells limit tissue destruction and facilitate repair within the murine CNS by clearance of cellular debris during EAE. TREM2 is a new attractive target for promotion of repair and resolution of inflammation in multiple sclerosis and other neuroinflammatory diseases.
Shigella flexneri, the pathogen responsible for bacillary dysentery, has evolved multiple strategies to control the inflammatory response. Here, we show that Shigella subverts the subcellular trafficking of the intercellular adhesion molecule-1 (ICAM-1), a key molecule in immune cell recruitment, in a mechanism dependent on the injected bacterial enzyme IpgD and its product, the lipid mediator PI5P. Overexpression of IpgD, but not a phosphatase dead mutant, induced the internalization and the degradation of ICAM-1 in intestinal epithelial cells. Remarkably, addition of permeant PI5P reproduced IpgD effects and led to the inhibition of neutrophil recruitment. Finally, these results were confirmed in an in vivo model of Shigella infection where IpgD-dependent ICAM-1 internalization reduced neutrophil adhesion. In conclusion, we describe here an immune evasion mechanism used by the pathogen Shigella to divert the host cell trafficking machinery in order to reduce immune cell recruitment.
Peroxiredoxin 6 (PRDX6) is one of the six members of the PRDX family, which have peroxidase and antioxidant activity. PRDX6 is unique, containing only one conserved cysteine residue (C47) rather than the two found in other PRDXs. A yeast two-hybrid screen found PRDX6 to be a potential binding partner of the C-terminal tail of anion exchanger 1 (AE1), a Cl(-)/HCO(3)(-) exchanger basolaterally expressed in renal a-intercalated cells. PRDX6 immunostaining in human kidney was both cytoplasmic and peripheral and colocalized with AE1. Analysis of native protein showed that it was largely monomeric, whereas expressed tagged protein was more dimeric. Two methionine oxidation sites were identified. In vitro and ex vivo pull-downs and immunoprecipitation assays confirmed interaction with AE1, but mutation of the conserved cysteine resulted in loss of interaction. Prdx6 knockout mice had a baseline acidosis with a major respiratory component and greater AE1 expression than wild-type animals. After an oral acid challenge, PRDX6 expression increased in wild-type mice, with preservation of AE1. However, AE1 expression was significantly decreased in knockout animals. Kidneys from acidified mice showed widespread proximal tubular vacuolation in wild-type but not knockout animals. Knockdown of PRDX6 by siRNA in mammalian cells reduced both total and cell membrane AE1 levels. Thus, PRDX6-AE1 interaction contributes to the maintenance of AE1 during cellular stress such as during metabolic acidosis.
Defective hepatic insulin receptor (IR) signalling is a pathogenic manifestation of metabolic disorders including obesity and diabetes. The endo/lysosomal trafficking system may coordinate insulin action and nutrient homeostasis by endocytosis of IR and the autophagic control of intracellular nutrient levels. Here we show that class III PI3K--a master regulator of endocytosis, endosomal sorting and autophagy--provides negative feedback on hepatic insulin signalling. The ultraviolet radiation resistance-associated gene protein (UVRAG)-associated class III PI3K complex interacts with IR and is stimulated by insulin treatment. Acute and chronic depletion of hepatic Vps15, the regulatory subunit of class III PI3K, increases insulin sensitivity and Akt signalling, an effect that requires functional IR. This is reflected by FoxO1-dependent transcriptional defects and blunted gluconeogenesis in Vps15 mutant cells. On depletion of Vps15, the metabolic syndrome in genetic and diet-induced models of insulin resistance and diabetes is alleviated. Thus, feedback regulation of IR trafficking and function by class III PI3K may be a therapeutic target in metabolic conditions of insulin resistance.
Microglia are tissue macrophages of the central nervous system (CNS) that control tissue homeostasis. Microglia dysregulation is thought to be causal for a group of neuropsychiatric, neurodegenerative and neuroinflammatory diseases, called "microgliopathies". However, how the intracellular stimulation machinery in microglia is controlled is poorly understood. Here, we identified the ubiquitin-specific protease (Usp) 18 in white matter microglia that essentially contributes to microglial quiescence. We further found that microglial Usp18 negatively regulates the activation of Stat1 and concomitant induction of interferon-induced genes, thereby terminating IFN signaling. The Usp18-mediated control was independent from its catalytic activity but instead required the interaction with Ifnar2. Additionally, the absence of Ifnar1 restored microglial activation, indicating a tonic IFN signal which needs to be negatively controlled by Usp18 under non-diseased conditions. These results identify Usp18 as a critical negative regulator of microglia activation and demonstrate a protective role of Usp18 for microglia function by regulating the Ifnar pathway. The findings establish Usp18 as a new molecule preventing destructive microgliopathy.
Macro-autophagy is a major catabolic process in the cell used to degrade protein aggregates, dysfunctional organelles and intracellular pathogens that would otherwise become toxic. Autophagy also generates energy and metabolites for the cell through recycling of degraded autophagosomal cargo, which can be particularly important for cell viability under stress. The significance of changes in the rates of autophagic flux for cellular function and disease is being increasingly appreciated, and interest in measuring autophagy in different experimental systems is growing accordingly. Here, we describe key methodologies used in the field to measure autophagic flux, including monitoring LC3 processing by western blot, fluorescent cell staining, and flow cytometry, in addition to changes in the levels or posttranslational modifications of other autophagy markers, such as p62/Sqstm1 and the Atg5-Atg12 conjugate. We also describe what cellular stresses may be used to induce autophagy and how to control for changes in the rates of autophagic flux as opposed to inhibition of flux. Finally, we detail available techniques to monitor autophagy in vivo.
Genetic studies have shown that the tuberous sclerosis complex (TSC) 1-TSC2-mammalian target of Rapamycin (mTOR) and the Hippo-Yes-associated protein 1 (YAP) pathways are master regulators of organ size, which are often involved in tumorigenesis. The crosstalk between these signal transduction pathways in coordinating environmental cues, such as nutritional status and mechanical constraints, is crucial for tissue growth. Whether and how mTOR regulates YAP remains elusive. Here we describe a novel mouse model of TSC which develops renal mesenchymal lesions recapitulating human perivascular epithelioid cell tumors (PEComas) from patients with TSC. We identify that YAP is up-regulated by mTOR in mouse and human PEComas. YAP inhibition blunts abnormal proliferation and induces apoptosis of TSC1-TSC2-deficient cells, both in culture and in mosaic Tsc1 mutant mice. We further delineate that YAP accumulation in TSC1/TSC2-deficient cells is due to impaired degradation of the protein by the autophagosome/lysosome system. Thus, the regulation of YAP by mTOR and autophagy is a novel mechanism of growth control, matching YAP activity with nutrient availability under growth-permissive conditions. YAP may serve as a potential therapeutic target for TSC and other diseases with dysregulated mTOR activity.
Pompe disease is a systemic metabolic disorder characterized by lack of acid-alpha glucosidase (GAA) resulting in ubiquitous lysosomal glycogen accumulation. Respiratory and ambulatory dysfunction are prominent features in patients with Pompe yet the mechanism defining the development of muscle weakness is currently unclear. Transgenic animal models of Pompe disease mirroring the patient phenotype have been invaluable in mechanistic and therapeutic study. Here, we demonstrate significant pathological alterations at neuromuscular junctions (NMJs) of the diaphragm and tibialis anterior muscle as prominent features of disease pathology in Gaa knockout mice. Postsynaptic defects including increased motor endplate area and fragmentation were readily observed in Gaa(-/-) but not wild-type mice. Presynaptic neuropathic changes were also evident, as demonstrated by significant reduction in the levels of neurofilament proteins, and alterations in axonal fiber diameter and myelin thickness within the sciatic and phrenic nerves. Our data suggest the loss of NMJ integrity is a primary contributor to the decline in respiratory and ambulatory function in Pompe and arises from both pre- and postsynaptic pathology. These observations highlight the importance of systemic phenotype correction, specifically restoration of GAA to skeletal muscle and the nervous system for treatment of Pompe disease.
The phosphatidylinositol 3-kinase (PI3K) regulatory subunits p55a and p50a are coordinately transcriptionally upregulated by signal transducer and activator of transcription 3 (Stat3) at the onset of mammary gland involution, a process that requires Stat3. Deletion of both p55a and p50a subunits in vivo abrogated mammary epithelial cell death during involution. This was associated also with reduced cytosolic levels and activity of the cysteine protease cathepsin L, which is implicated in lysosomal-mediated programmed cell death (LM-PCD) and is upregulated in involution. Furthermore, involution is delayed in cathepsin L-deficient mice suggesting that the p55a/p50a subunits mediate cell death in part by elevating the level of cathepsin L resulting in increased cytosolic activity. Surprisingly, we found that p55a/p50a localize to the nucleus where they bind to chromatin and regulate transcription of a subset of inflammatory/acute phase genes that are also Stat3 targets. Our findings reveal a novel role for these PI3K regulatory subunits as regulators of LM-PCD in vivo.
Macroautophagy is a highly conserved intracellular process responsible for the degradation of subcellular constituents. Macroautophagy was recently suggested to be involved in the removal of mitochondria from reticulocytes during the final stage of erythrocyte differentiation. Although Atg5 and Atg7 are indispensable for macroautophagy, their role in mitochondrial clearance remains controversial. We recently discovered that mammalian cells use conventional Atg5/Atg7-dependent macroautophagy as well as an alternative Unc-51-like kinase 1 (Ulk1)-dependent Atg5/Atg7-independent macroautophagy process. We hypothesized that the latter may be involved in mitochondrial clearance from reticulocytes during erythrocyte differentiation. Here we report that fetal definitive reticulocytes from Ulk1-deficient and Ulk1/Atg5 double-deficient mice retain their mitochondria, whereas the mitochondria are engulfed and digested within autophagic structures in wild-type and Atg5-deficient mice. Mitochondrial retention by Ulk1-deficient reticulocytes is far less marked in primitive and adult definitive reticulocytes. These data indicate that Ulk1-dependent Atg5-independent macroautophagy is the dominant process of mitochondrial clearance from fetal definitive reticulocytes.
Microglia are brain macrophages and, as such, key immune-competent cells that can respond to environmental changes. Understanding the mechanisms of microglia-specific responses during pathologies is hence vital for reducing disease burden. The definition of microglial functions has so far been hampered by the lack of genetic in vivo approaches that allow discrimination of microglia from closely related peripheral macrophage populations in the body. Here we introduce a mouse experimental system that specifically targets microglia to examine the role of a mitogen-associated protein kinase kinase kinase (MAP3K), transforming growth factor (TGF)-ß-activated kinase 1 (TAK1), during autoimmune inflammation. Conditional depletion of TAK1 in microglia only, not in neuroectodermal cells, suppressed disease, significantly reduced CNS inflammation and diminished axonal and myelin damage by cell-autonomous inhibition of the NF-?B, JNK and ERK1/2 pathways. Thus, we found TAK1 to be pivotal in CNS autoimmunity, and we present a tool for future investigations of microglial function in the CNS.
Microglia are crucial for immune responses in the brain. Although their origin from the yolk sac has been recognized for some time, their precise precursors and the transcription program that is used are not known. We found that mouse microglia were derived from primitive c-kit(+) erythromyeloid precursors that were detected in the yolk sac as early as 8 d post conception. These precursors developed into CD45(+) c-kit(lo) CX(3)CR1(-) immature (A1) cells and matured into CD45(+) c-kit(-) CX(3)CR1(+) (A2) cells, as evidenced by the downregulation of CD31 and concomitant upregulation of F4/80 and macrophage colony stimulating factor receptor (MCSF-R). Proliferating A2 cells became microglia and invaded the developing brain using specific matrix metalloproteinases. Notably, microgliogenesis was not only dependent on the transcription factor Pu.1 (also known as Sfpi), but also required Irf8, which was vital for the development of the A2 population, whereas Myb, Id2, Batf3 and Klf4 were not required. Our data provide cellular and molecular insights into the origin and development of microglia.
Niemann-Pick disease (NPD) is a lysosomal storage disease caused by the loss of acid sphingomyelinase (ASMase) that features neurodegeneration and liver disease. Because ASMase-knock-out mice models NPD and our previous findings revealed that ASMase activates cathepsins B/D (CtsB/D), our aim was to investigate the expression and processing of CtsB/D in hepatic stellate cells (HSCs) from ASMase-null mice and their role in liver fibrosis. Surprisingly, HSCs from ASMase-knock-out mice exhibit increased basal level and activity of CtsB as well as its in vitro processing in culture, paralleling the enhanced expression of fibrogenic markers a-smooth muscle actin (a-SMA), TGF-ß, and pro-collagen-a1(I) (Col1A1). Moreover, pharmacological inhibition of CtsB blunted the expression of a-SMA and Col1A1 and proliferation of HSCs from ASMase-knock-out mice. Consistent with the enhanced activation of CtsB in HSCs from ASMase-null mice, the in vivo liver fibrosis induced by chronic treatment with CCl(4) increased in ASMase-null compared with wild-type mice, an effect that was reduced upon CtsB inhibition. In addition to liver, the enhanced proteolytic processing of CtsB was also observed in brain and lung of ASMase-knock-out mice, suggesting that the overexpression of CtsB may underlie the phenotype of NPD. Thus, these findings reveal a functional relationship between ASMase and CtsB and that the ablation of ASMase leads to the enhanced processing and activation of CtsB. Therefore, targeting CtsB may be of relevance in the treatment of liver fibrosis in patients with NPD.
It is well established that lysosomes play an active role during the execution of cell death. A range of stimuli can lead to lysosomal membrane permeabilization (LMP), thus inducing programmed cell death without involvement of the classical apoptotic programme. However, these lysosomal pathways of cell death have mostly been described in vitro or under pathological conditions. Here we show that the physiological process of post-lactational regression of the mammary gland is accomplished through a non-classical, lysosomal-mediated pathway of cell death. We found that, during involution, lysosomes in the mammary epithelium undergo widespread LMP. Furthermore, although cell death through LMP is independent of executioner caspases 3, 6 and 7, it requires Stat3, which upregulates the expression of lysosomal proteases cathepsin B and L, while downregulating their endogenous inhibitor Spi2A (ref. 8). Our findings report a previously unknown, Stat3-regulated lysosomal-mediated pathway of cell death under physiological circumstances. We anticipate that these findings will be of major importance in the design of treatments for cancers such as breast, colon and liver, where cathepsins and Stat3 are commonly overexpressed and/or hyperactivated respectively.
lgp110 is a heavily glycosylated intrinsic protein of lysosomal membranes. Initially defined by monoclonal antibodies against mouse liver lysosomes, it consists of a 45-kilodalton core polypeptide with O-linked and 17 asparagine-linked oligosaccharide side chains in mouse cells. Sialic acid residues make the mature protein extremely acidic, with an isoelectric point of between 2 and 4 in both normal tissues and most cultured cell lines. Partial sequencing of mouse lgp110 allowed oligonucleotide probes to be constructed for the screening of several mouse cDNA libraries. A partial cDNA clone for mouse lgp110 was found and used for additional library screening, generating a cDNA clone covering all of the coding sequence of mature rat lgp110 as well as genomic clones covering most of the mouse gene. These new clones bring to seven the number of lysosomal membrane proteins whose amino acid sequences can be deduced, and two distinct but highly similar groups (designated lgp-A and lgp-B) can now be defined. Sequence comparisons suggest that differences within each group reflect species variations of the same protein and that lgp-A and lgp-B probably diverged from a common ancestor prior to the evolup4f1ary divergence of birds and mammals. Individual cells and individual lysosomes possess both lgp-A and lgp-B, suggesting that these two proteins have different functions. Mouse lgp110 is encoded by at least seven exons; intron positions suggest that the two homologous ectodomains of each lgp arose through gene duplication.
Background: Rotenone is an environmental neurotoxin that induces accumulation of a-synuclein and degeneration of dopaminergic neurons in substantia nigra pars compacta (SNpc), but the molecular mechanisms are not fully understood. We investigated whether rotenone induced impairment of autophagic flux and lysosomal functions.
Methods: Autophagy flux, accumulation of a-synuclein, lysosomal membrane integrity and neurodegeneration were assessed in the rotenone-treated rat model and PC12 cells, and the effects of the autophagy inducer trehalose on rotenone's cytotoxicity were also studied.
Results: Rotenone administration significantly reduced motor activity and caused a loss of tyrosine hydroxylase in SNpc of Lewis rats. The degeneration of nigral dopaminergic neurons was accompanied by the deposition of a-synuclein aggregates, autophagosomes and redistribution of cathepsin D from lysosomes to the cytosol. In cultured PC12 cells, rotenone also induced increases in protein levels of a-synuclein, microtubule-associated protein 1 light chain 3-II, Beclin 1, and p62. Rotenone increased lysosomal membrane permeability as evidenced by leakage of N-acetyl-beta-d-glucosaminidase and cathepsin D, the effects were blocked by reactive oxygen species scavenger tiron. Autophagy inducer trehalose enhanced the nuclear translocation of transcription factor EB, accelerated the clearance of autophagosomes and a-synuclein and attenuated rotenone-induced cell death of PC12 cells. Meanwhile, administration of trehalose to rats in drinking water (2%) decreased rotenone-induced dopaminergic neurons loss in SNpc.
Conclusions: These studies indicate that the lysosomal dysfunction contributes to rotenone's neurotoxicity and restoration of lysosomal function could be a new therapeutic strategy for Parkinson's disease.
Objective: Globotriaosylceramide (Gb3) induces KCa3.1 downregulation in Fabry disease (FD). We investigated whether Gb3 induces KCa3.1 endocytosis and degradation.
Approach and results: KCa3.1, especially plasma membrane-localized KCa3.1, was downregulated in both Gb3-treated mouse aortic endothelial cells (MAECs) and human umbilical vein endothelial cells. Gb3-induced KCa3.1 downregulation was prevented by lysosomal inhibitors but not by a proteosomal inhibitor. Endoplasmic reticulum stress-inducing agents did not induce KCa3.1 downregulation. Gb3 upregulated the protein levels of early endosome antigen 1 and lysosomal-associated membrane protein 2 in MAECs. Compared with MAECs from age-matched wild-type mice, those from aged a-galactosidase A (Gla)-knockout mice, an animal model of FD, showed downregulated KCa3.1 expression and upregulated early endosome antigen 1 and lysosomal-associated membrane protein 2 expression. In contrast, no significant difference was found in early endosome antigen 1 and lysosomal-associated membrane protein 2 expression between young Gla-knockout and wild-type MAECs. In aged Gla-knockout MAECs, clathrin was translocated close to the cell border and clathrin knockdown recovered KCa3.1 expression. Rab5, an effector of early endosome antigen 1, was upregulated, and Rab5 knockdown restored KCa3.1 expression, the current, and endothelium-dependent relaxation.
Conclusions: -Gb3 accelerates the endocytosis and lysosomal degradation of endothelial KCa3.1 via a clathrin-dependent process, leading to endothelial dysfunction in FD.

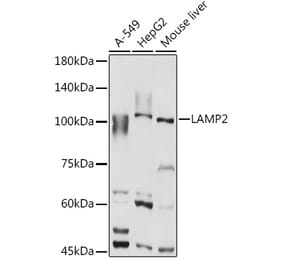
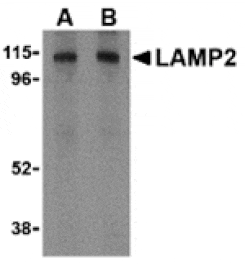
![Western Blot - Anti-LAMP2 Antibody [ARC0274] (A305918) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305918_1.jpg?profile=product_alternative)


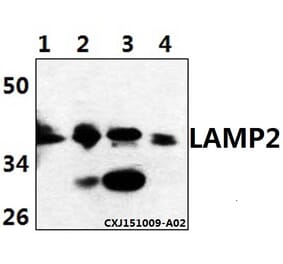
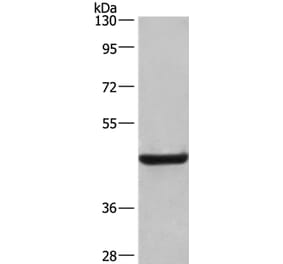
![Western Blot - Anti-LAMP2 Antibody [GL2A7] (A304947) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304947_1.png?profile=product_top)
![Immunocytochemistry/Immunofluorescence - Anti-LAMP2 Antibody [GL2A7] (A304947) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304947_2.png?profile=product_top)
![Western Blot - Anti-LAMP2 Antibody [GL2A7] (A304947) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304947_1.png?profile=product_top_thumb)
![Immunocytochemistry/Immunofluorescence - Anti-LAMP2 Antibody [GL2A7] (A304947) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304947_2.png?profile=product_top_thumb)
![Western Blot - Anti-LAMP2 Antibody [GL2A7] (A304947) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304947_1.png?profile=product_image)
![Immunocytochemistry/Immunofluorescence - Anti-LAMP2 Antibody [GL2A7] (A304947) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304947_2.png?profile=product_image)