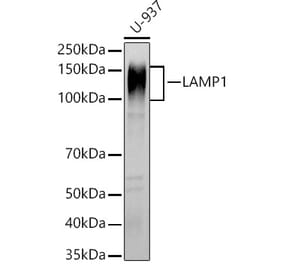
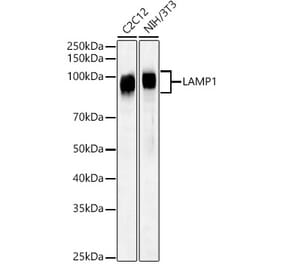
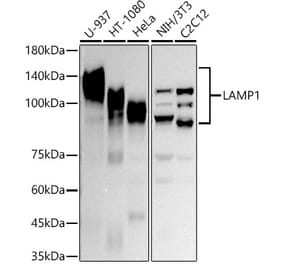
Western Blot - Anti-LAMP1 Antibody [Ly1C6] (A305123)
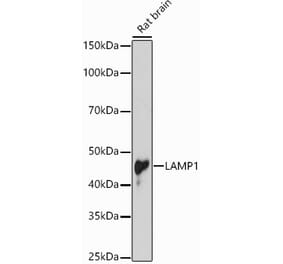
Western blot analysis of rat liver microsome lysate showing detection of LAMP1 protein using Anti-LAMP1 Antibody [Ly1C6] (A305123) at 1:1,000 for 2 hours at room temperature. Load: 15 µg. Block: 1.5% BSA for 30 minutes at room temperature. The secondary antibody used was Sheep Anti-Mouse IgG: HRP for 1 hour at room temperature.
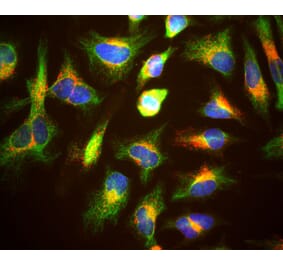
Immunocytochemistry/Immunofluorescence analysis of human transfected HeLa cells, using Anti-LAMP1 Antibody [Ly1C6] (A305123), at 1:1,000. The secondary antibody used was APC Goat Anti-Mouse (red).
Publishing research using Anti-LAMP1 Antibody [Ly1C6] (A305123)? Please let us know so that we can list the citation on this page.


![Western Blot - Anti-LAMP1 Antibody [Ly1C6] (A305123) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305123_1.png?profile=product_top)
![Immunocytochemistry/Immunofluorescence - Anti-LAMP1 Antibody [Ly1C6] (A305123) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305123_2.png?profile=product_top)
![Western Blot - Anti-LAMP1 Antibody [Ly1C6] (A305123) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305123_1.png?profile=product_top_thumb)
![Immunocytochemistry/Immunofluorescence - Anti-LAMP1 Antibody [Ly1C6] (A305123) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305123_2.png?profile=product_top_thumb)
![Western Blot - Anti-LAMP1 Antibody [Ly1C6] (A305123) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305123_1.png?profile=product_image)
![Immunocytochemistry/Immunofluorescence - Anti-LAMP1 Antibody [Ly1C6] (A305123) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305123_2.png?profile=product_image)
![Flow Cytometry - Anti-CD107a Antibody [H4A3] - BSA and Azide free (A86536) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86537_711.jpg?profile=product_alternative)

![SDS-PAGE - Anti-LAMP1 Antibody [SAR428926] - Low endotoxin, Azide free (A324125) - Antibodies.com](https://cdn.antibodies.com/image/catalog/324/A324125_1.jpg?profile=product_alternative)