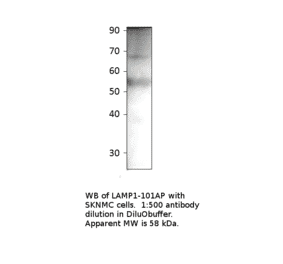
Anti-LAMP1 Antibody [H4A3] - BSA and Azide free (A86537) has been discontinued and is no longer available.
View all LAMP1 Antibodies.
Unconjugated


![Flow Cytometry - Anti-CD107a Antibody [H4A3] - BSA and Azide free (A86536) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86537_711.jpg?profile=product_top)
![Flow Cytometry - Anti-CD107a Antibody [H4A3] - BSA and Azide free (A86536) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86537_711.jpg?profile=product_top_thumb)
![Flow Cytometry - Anti-CD107a Antibody [H4A3] - BSA and Azide free (A86536) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86537_711.jpg?profile=product_image)
![Immunofluorescence - Anti-LAMP1 Antibody [5H6] (A85308) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85308_1.jpg?profile=product_alternative)
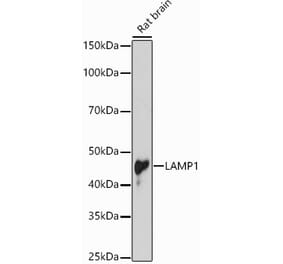
![Western Blot - Anti-LAMP1 Antibody [Ly1C6] (A305123) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305123_1.png?profile=product_alternative)

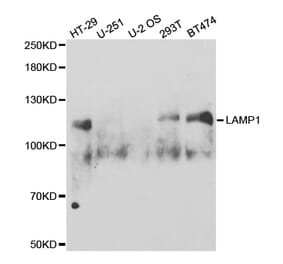
![SDS-PAGE - Anti-LAMP1 Antibody [SAR428926] - Low endotoxin, Azide free (A324125) - Antibodies.com](https://cdn.antibodies.com/image/catalog/324/A324125_1.jpg?profile=product_alternative)
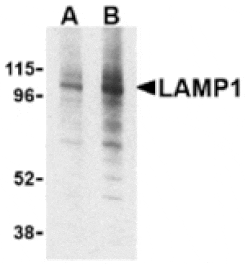
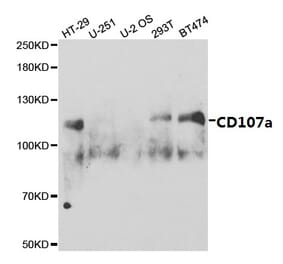
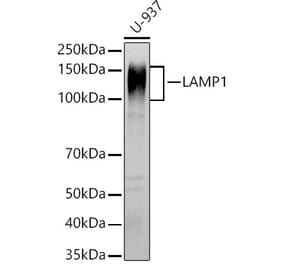
![SDS-PAGE - Anti-LAMP1 Antibody [SAR428926] Biosimilar - BSA and Azide free (A340105) - Antibodies.com](https://cdn.antibodies.com/image/catalog/340/A340105_1.jpg?profile=product_alternative)