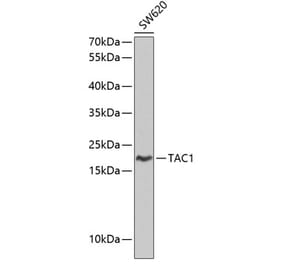
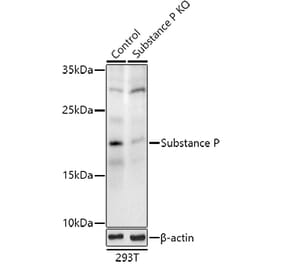
Unconjugated
ß-arrestin2 is a key molecule involved in signaling and internalization of activated G protein-coupled receptors including µ-opioid receptors (MOR). Previously we have shown that decreased expression of ß-arrestin2 upon chronic morphine is associated with the development of opioid tolerance in the gastrointestinal tract. However, the localization of ß-arrestin2 within the gastrointestinal wall is not known. In this study we found that ß-arrestin2 is localized in the soma of a select group of neurons in the myenteric ganglia but not in smooth muscle. The density of ß-arestin2 was significantly higher in the ileum than the colon. We identified four variants of ß-arrestin2 in the ileum, with ARRB-005 and ARRB-013 being the most abundant. Further, the current study utilized multiple-labeling immunofluorescence to characterize the chemical coding of neurons expressing ß-arrestin2 in the murine myenteric plexus and the co-localization of MOR1 and ß-arrestin2. ß-arrestin2 co-localized with choline acetyltransferase and calretinin. In contrast, ß-arrestin2 neither co-localized with substance P, nitric oxide synthase nor calbindin. Genetic deletion of ß-arrestin2 did not affect cholinergic neuron activation by nicotine in the isolated ileum (-log M EC50: wild type?=?5.8 vs. ß-arrestin2 knockout?=?5.9). Our findings suggest specificity in the localization of ß-arrestin2 in the myenteric plexus within MOR1-expressing neurons and provide a relation for direct intracellular crosstalk between MOR1 receptor activation and ß-arrestin2 signaling in the myenteric neurons. ß-arrestin2 deletion does not directly alter basal enteric cholinergic neuronal function.
Children are uniquely susceptible to ozone because airway and lung growth continue for an extensive period after birth. Early-life exposure of the rhesus monkey to repeated ozone cycles results in region-specific disrupted airway/lung growth, but the mediators and mechanisms are poorly understood. Substance P (SP), neurokinin-1 receptor (NK-1R); and nuclear receptor Nur77 (NR4A1) are signaling pathway components involved in ozone-induced cell death. We hypothesize that acute ozone (AO) exposure during postnatal airway development disrupts SP/NK-1R/Nur77 pathway expression and that these changes correlate with increased ozone-induced cell death. Our objectives were to 1) spatially define the normal development of the SP/NK-1R/Nur77 pathway in conducting airways; 2) compare how postnatal age modulates responses to AO exposure; and 3) determine how concomitant, episodic ozone exposure modifies age-specific acute responses. Male infant rhesus monkeys were assigned at age 1 mo to two age groups, 2 or 6 mo, and then to one of three exposure subgroups: filtered air (FA), FA+AO (AO: 8 h/day × 2 days), or episodic biweekly ozone exposure cycles (EAO: 8 h/day × 5 days/14-day cycle+AO). O3 = 0.5 ppm. We found that 1) ozone increases SP/NK-1R/Nur77 pathway expression in conducting airways, 2) an ozone exposure cycle (5 days/cycle) delivered early at age 2 mo resulted in an airway that was hypersensitive to AO exposure at the end of 2 mo, and 3) continued episodic exposure (11 cycles) resulted in an airway that was hyposensitive to AO exposure at 6 mo. These observations collectively associate with greater overall inflammation and epithelial cell death, particularly in early postnatal (2 mo), distal airways.
Copyright © 2014 the American Physiological Society.