Explore our range of 10 FGFR1 ELISA kits for the qualitative or quantitative detection of FGFR1 from human, mouse and rat samples.
Synonyms: Basic fibroblast growth factor receptor 1, bFGF-R-1, BFGFR, CD331, CEK, FGFBR, FGFR-1, Fibroblast growth factor receptor 1, FLG, FLT-2, FLT2, Fms-like tyrosine kinase 2, HBGFR, N-sam and Proto-oncogene c-Fgr
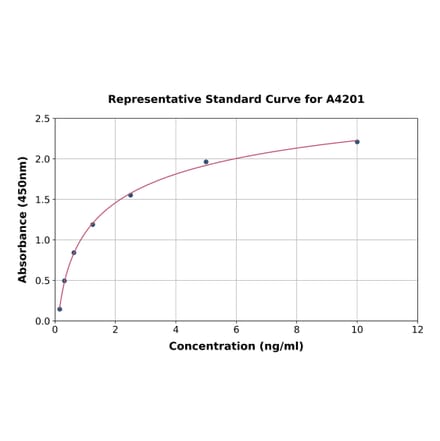
Human FGFR1 ELISA Kit (A4201) | |
48T - 96T/$550 – $695 | |
| Applications: | Sandwich ELISA |
| Reactivity: | Human |
| Sample Type: | Tissue homogenates, cell lysates or other biological fluids. |
| Range: | 0.156-10 ng/ml |
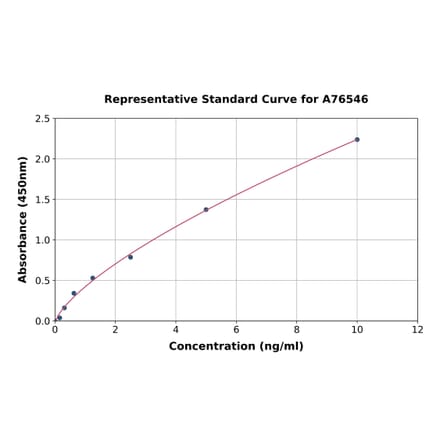
Human FGFR1 ELISA Kit (A76546) | |
96T/$625 | |
| Applications: | Sandwich ELISA |
| Reactivity: | Human |
| Sample Type: | Serum, plasma, tissue homogenates, and other biological fluids. |
| Range: | 0.156-10 ng/ml |
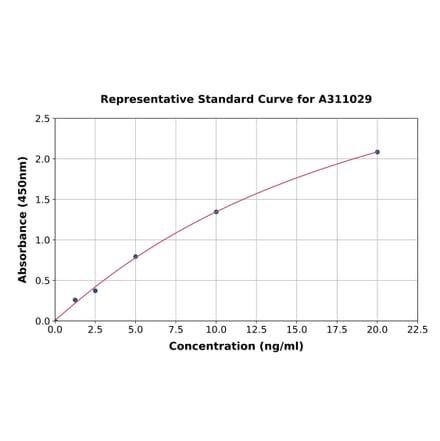
Human FGFR1 ELISA Kit (A311029) | |
96T/$545 | |
| Applications: | Sandwich ELISA |
| Reactivity: | Human |
| Sample Type: | Serum, plasma or other biological fluids. |
| Range: | 1.25-20 ng/ml |
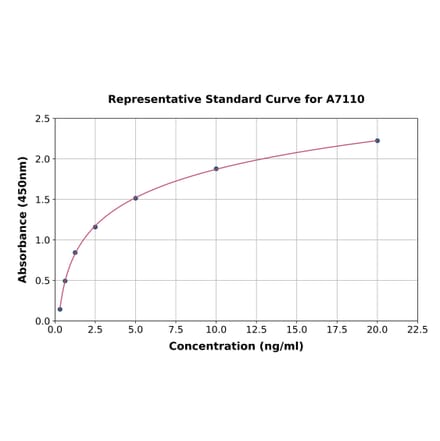
Rat FGFR1 ELISA Kit (A7110) | |
48T - 96T/$575 – $740 | |
| Applications: | Sandwich ELISA |
| Reactivity: | Rat |
| Sample Type: | Tissue homogenates, cell lysates or other biological fluids. |
| Range: | 0.312-20 ng/ml |
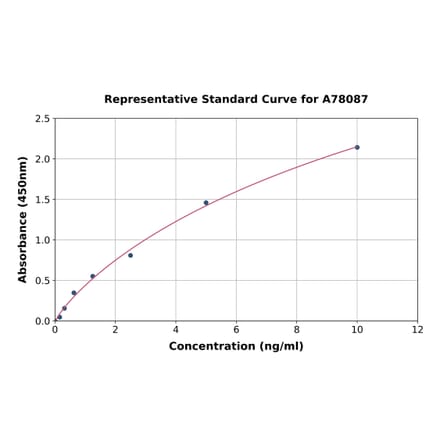
Rat FGFR1 ELISA Kit (A78087) | |
96T/$625 | |
| Applications: | Sandwich ELISA |
| Reactivity: | Rat |
| Sample Type: | Serum, plasma, tissue homogenates, and other biological fluids. |
| Range: | 0.156-10 ng/ml |
Mouse FGFR1 ELISA Kit (A4401) | |
48T - 96T/$560 – $710 | |
| Applications: | Sandwich ELISA |
| Reactivity: | Mouse |
| Sample Type: | Tissue homogenates or other biological fluids. |
| Range: | 0.156-10 ng/ml |
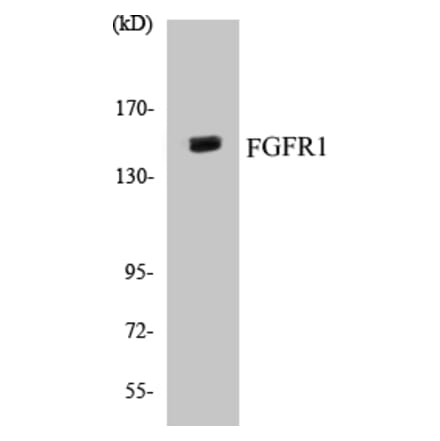
FGFR1 Cell Based ELISA Kit (A103041) | |
96T/$570 | |
| Reactivity: | Human, Mouse, Rat |
| Sample Type: | Adherent cells and suspension cells. |
| Range: | > 5000 Cells |
| Detection Type: | Colorimetric |
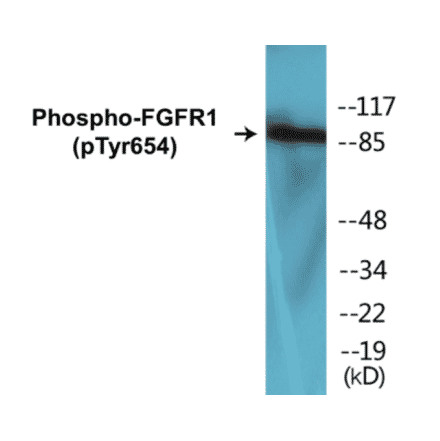
FGFR1 (phospho Tyr654) Cell Based ELISA Kit (A102131) | |
2 x 96T/$670 | |
| Reactivity: | Human, Mouse, Rat |
| Sample Type: | Adherent cells and suspension cells. |
| Range: | > 5000 Cells |
| Detection Type: | Colorimetric |
FGFR1 (phospho Tyr154) Cell Based ELISA Kit (A102199) | |
2 x 96T/$670 | |
| Reactivity: | Human, Mouse, Rat |
| Sample Type: | Adherent cells and suspension cells. |
| Range: | > 5000 Cells |
| Detection Type: | Colorimetric |
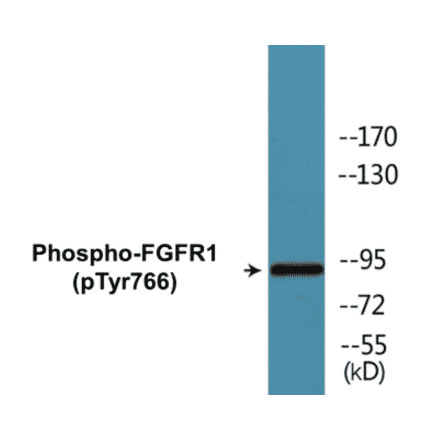
FGFR1 (phospho Tyr766) Cell Based ELISA Kit (A102152) | |
2 x 96T/$670 | |
| Reactivity: | Human, Mouse, Rat |
| Sample Type: | Adherent cells and suspension cells. |
| Range: | > 5000 Cells |
| Detection Type: | Colorimetric |
Showing 1-10 of 10 products