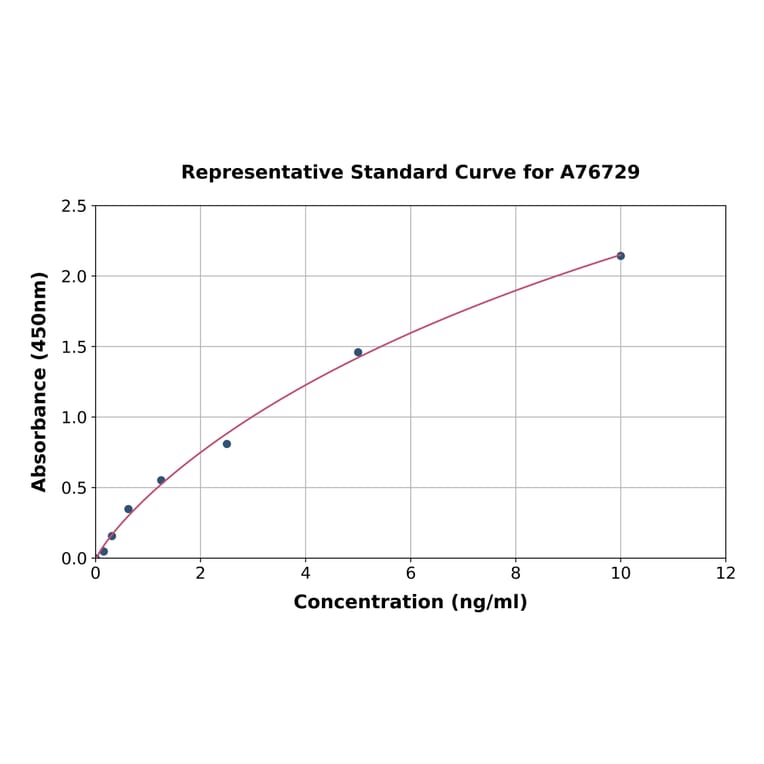
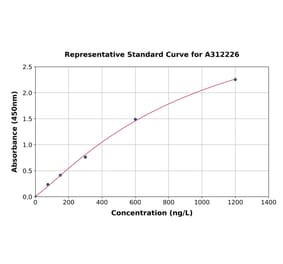
| Sample Type | n | Range | Average |
|---|---|---|---|
| Serum | 5 | 86% - 98% | 93% |
| EDTA Plasma | 5 | 87% - 104% | 94% |
| Heparin Plasma | 5 | 87% - 104% | 92% |
| Sample Type | n | 1:2 | 1:4 | 1:8 |
|---|---|---|---|---|
| Serum | 5 | 88-105% | 87-101% | 90-105% |
| EDTA Plasma | 5 | 91-101% | 87-94% | 85-98% |
| Heparin Plasma | 5 | 85-98% | 81-97% | 83-98% |
| Item | Quantity | Storage |
|---|---|---|
| Pre-Coated 96 Well Microplate | 12 x 8 Well Strips | +4°C |
| Lyopholized Standard | 2 Vials | +4°C |
| Sample Dilution Buffer | 20ml | +4°C |
| Biotinylated Detection Antibody | 120µl | +4°C |
| Antibody Dilution Buffer | 10ml | +4°C |
| HRP-Streptavidin Conjugate | 120µl | +4°C |
| SABC Dilution Buffer | 10ml | +4°C |
| TMB Substrate | 10ml | +4°C |
| Stop Solution | 10ml | +4°C |
| Wash Buffer (25X) | 30ml | +4°C |
| Plate Sealers | 5 Adhesive Strips | - |
| Foil Pouch | 1 Zip-Sealed Pouch | - |
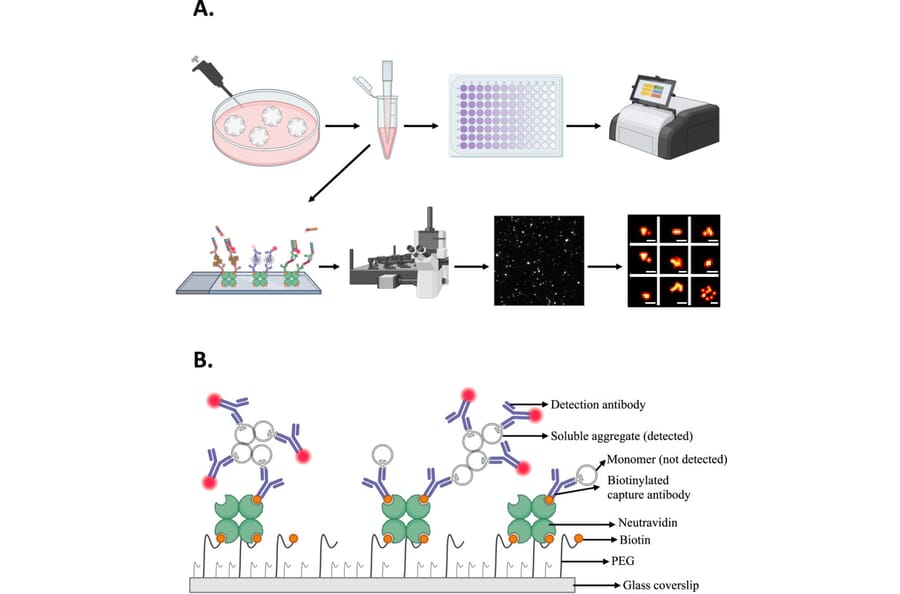
Understanding the role of small, soluble aggregates of beta-amyloid (Aß) and tau in Alzheimer's disease (AD) is of great importance for the rational design of preventative therapies. Here we report a set of methods for the detection, quantification, and characterisation of soluble aggregates in conditioned media of cerebral organoids derived from human iPSCs with trisomy 21, thus containing an extra copy of the amyloid precursor protein (APP) gene. We detected soluble beta-amyloid (Aß) and tau aggregates secreted by cerebral organoids from both control and the isogenic trisomy 21 (T21) genotype. We developed a novel method to normalise measurements to the number of live neurons within organoid-conditioned media based on glucose consumption. Thus normalised, T21 organoids produced 2.5-fold more Aß aggregates with a higher proportion of larger (300-2000 nm2) and more fibrillary-shaped aggregates than controls, along with 1.3-fold more soluble phosphorylated tau (pTau) aggregates, increased inflammasome ASC-specks, and a higher level of oxidative stress inducing thioredoxin-interacting protein (TXNIP). Importantly, all this was detectable prior to the appearance of histological amyloid plaques or intraneuronal tau-pathology in organoid slices, demonstrating the feasibility to model the initial pathogenic mechanisms for AD in-vitro using cells from live genetically pre-disposed donors before the onset of clinical disease. Then, using different iPSC clones generated from the same donor at different times in two independent experiments, we tested the reproducibility of findings in organoids. While there were differences in rates of disease progression between the experiments, the disease mechanisms were conserved. Overall, our results show that it is possible to non-invasively follow the development of pathology in organoid models of AD over time, by monitoring changes in the aggregates and proteins in the conditioned media, and open possibilities to study the time-course of the key pathogenic processes taking place.