Unconjugated
This study presents human placenta-derived multipotent cells (PDMCs) as a source from which functional glutamatergic neurons can be derived. We found that the small heat-shock protein 27 (HSP27) was downregulated during the neuronal differentiation process. The in vivo temporal and spatial profiles of HSP27 expression were determined and showed inverted distributions with neuronal proteins during mouse embryonic development. Overexpression of HSP27 in stem cells led to the arrest of neuronal differentiation; however, the knockdown of HSP27 yielded a substantially enhanced ability of PDMCs to differentiate into neurons. These neurons formed synaptic networks and showed positive staining for multiple neuronal markers. Additionally, cellular phenomena including the absence of apoptosis and rare proliferation in HSP27-silenced PDMCs, combined with molecular events such as cleaved caspase-3 and the loss of stemness with cleaved Nanog, indicated that HSP27 is located upstream of neuronal differentiation and constrains that process. Furthermore, the induced neurons showed increasing intracellular calcium concentrations upon glutamate treatment. These differentiated cells co-expressed the N-methyl-D-aspartate receptor, vesicular glutamate transporter, and synaptosomal-associated protein 25 but did not show expression of tyrosine hydroxylase, choline acetyltransferase or glutamate decarboxylase 67. Therefore, we concluded that HSP27-silenced PDMCs differentiated into neurons possessing the characteristics of functional glutamatergic neurons.
Mutations in the C terminus of the serotonin transporter (SERT) disrupt folding and export from the endoplasmic reticulum. Here we examined the hypothesis that a cytosolic heat shock protein relay was recruited to the C terminus to assist folding of SERT. This conjecture was verified by the following observations. (i) The proximal portion of the SERT C terminus conforms to a canonical binding site for DnaK/heat shock protein of 70 kDa (HSP70). A peptide covering this segment stimulated ATPase activity of purified HSP70-1A. (ii) A GST fusion protein comprising the C terminus of SERT pulled down HSP70-1A. The interaction between HSP70-1A and SERT was visualized in live cells by Förster resonance energy transfer: it was restricted to endoplasmic reticulum-resident transporters and enhanced by an inhibitor that traps HSP70-1A in its closed state. (iv) Co-immunoprecipitation confirmed complex formation of SERT with HSP70-1A and HSP90ß. Consistent with an HSP relay, co-chaperones (e.g. HSC70-HSP90-organizing protein) were co-immunoprecipitated with the stalled mutants SERT-R607A/I608A and SERT-P601A/G602A. (v) Depletion of HSP90ß by siRNA or its inhibition increased the cell surface expression of wild type SERT and SERT-F604Q. In contrast, SERT-R607A/I608A and SERT-P601A/G602A were only rendered susceptible to inhibition of HSP70 and HSP90 by concomitant pharmacochaperoning with noribogaine. (vi) In JAR cells, inhibition of HSP90 also increased the levels of SERT, indicating that endogenously expressed transporter was also susceptible to control by HSP90ß. These findings support the concept that the folding trajectory of SERT is sampled by a cytoplasmic chaperone relay.
![FACS - Anti-HSP70 Antibody [C92F3A-5] (A304773) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304773_1.png?profile=product_top)
![Western Blot - Anti-HSP70 Antibody [C92F3A-5] (A304773) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304773_2.png?profile=product_top)
![Immunohistochemistry - Anti-HSP70 Antibody [C92F3A-5] (A304773) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304773_3.png?profile=product_top)
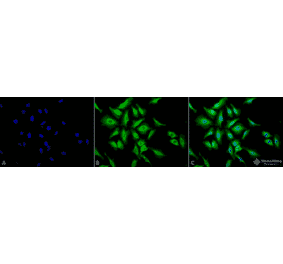
![Immunocytochemistry/Immunofluorescence - Anti-HSP70 Antibody [C92F3A-5] (A304773) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304773_4.png?profile=product_top)
![Immunohistochemistry - Anti-HSP70 Antibody [C92F3A-5] (A304773) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304773_5.png?profile=product_top)
![FACS - Anti-HSP70 Antibody [C92F3A-5] (A304773) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304773_1.png?profile=product_top_thumb)
![Western Blot - Anti-HSP70 Antibody [C92F3A-5] (A304773) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304773_2.png?profile=product_top_thumb)
![Immunohistochemistry - Anti-HSP70 Antibody [C92F3A-5] (A304773) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304773_3.png?profile=product_top_thumb)
![Immunocytochemistry/Immunofluorescence - Anti-HSP70 Antibody [C92F3A-5] (A304773) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304773_4.png?profile=product_top_thumb)
![Immunohistochemistry - Anti-HSP70 Antibody [C92F3A-5] (A304773) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304773_5.png?profile=product_top_thumb)
![FACS - Anti-HSP70 Antibody [C92F3A-5] (A304773) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304773_1.png?profile=product_image)
![Western Blot - Anti-HSP70 Antibody [C92F3A-5] (A304773) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304773_2.png?profile=product_image)
![Immunohistochemistry - Anti-HSP70 Antibody [C92F3A-5] (A304773) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304773_3.png?profile=product_image)
![Immunocytochemistry/Immunofluorescence - Anti-HSP70 Antibody [C92F3A-5] (A304773) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304773_4.png?profile=product_image)
![Immunohistochemistry - Anti-HSP70 Antibody [C92F3A-5] (A304773) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304773_5.png?profile=product_image)




















![Western Blot - Anti-HSP70 Antibody [N27F3-4] (A305087) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305087_1.png?profile=product_alternative)
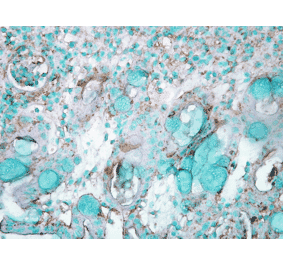
![Immunohistochemistry - Anti-HSP70 Antibody [BB70] (A305113) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305113_1.png?profile=product_alternative)
![Western Blot - Anti-HSP70 Antibody [3A3] (A305077) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305077_1.png?profile=product_alternative)
![Western Blot - Anti-HSP70 Antibody [5A5] (A305076) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305076_1.png?profile=product_alternative)
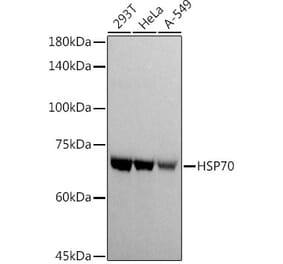
![Western Blot - Anti-HSP70 Antibody [2A4] (A305075) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305075_1.png?profile=product_alternative)
![FACS - Anti-HSP70 Antibody [1H11] (A304712) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304712_1.png?profile=product_alternative)
![Western Blot - Anti-HSP70 Antibody [1.86] (A304774) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304774_1.png?profile=product_alternative)