Recognizes the epitope of 6x His-tags encoded by many commercially available vectors, regardless of the tag's location in the fusion protein sequences (i.e. reacts with N-terminal, C-terminal or internal 6x His-tags).
Applications
Dot, ELISA, IP, IS, WB
Immunogen
HHHHHH (6x His) synthetic peptide conjugated to KLH.
Host
Mouse
Clonality
Monoclonal
Clone ID
HIS.H8
Isotype
IgG2b
Conjugate
Unconjugated
Purification
Protein A affinity chromatography from mouse ascites fluid.
Concentration
1 mg/ml
Product Form
Liquid
Formulation
Supplied in 10mM Phosphate Buffered Saline, pH 7.20, with 0.05% Sodium Azide.
Storage
Shipped at 4°C. Upon delivery aliquot and store at -20°C. Avoid freeze / thaw cycles.
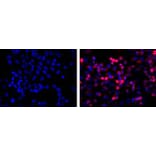
Immunofluorescence - Anti-His Tag Antibody [HIS.H8] (A85277)
Immunofluorescence (red) with Anti-His Tag Antibody on His-tag fusion protein in HEK293 cells. Left hand panel: untransfected control. Right hand panel: transfected.?Counterstained with DAPI (blue).
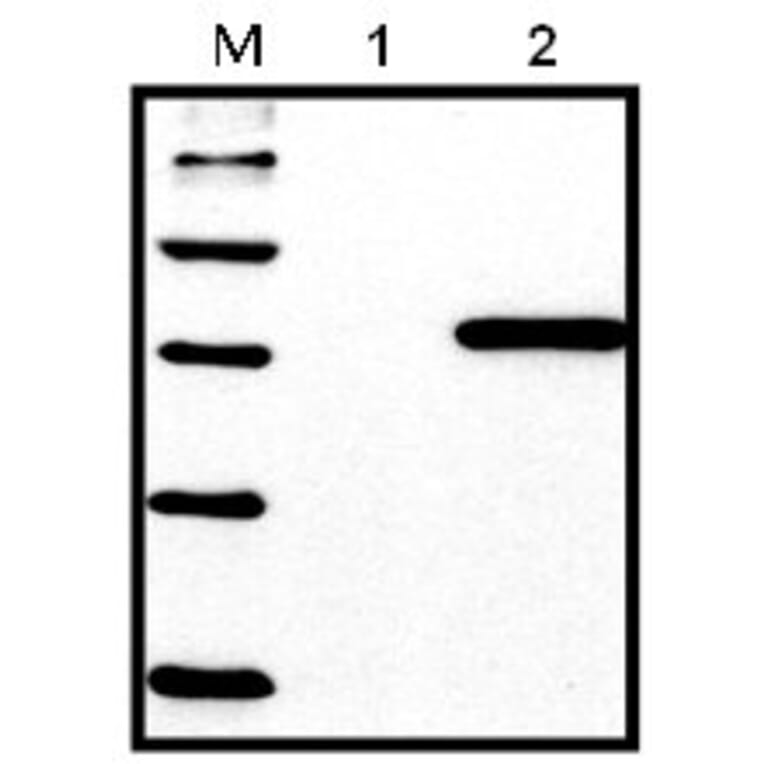
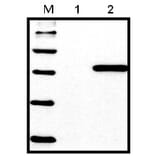
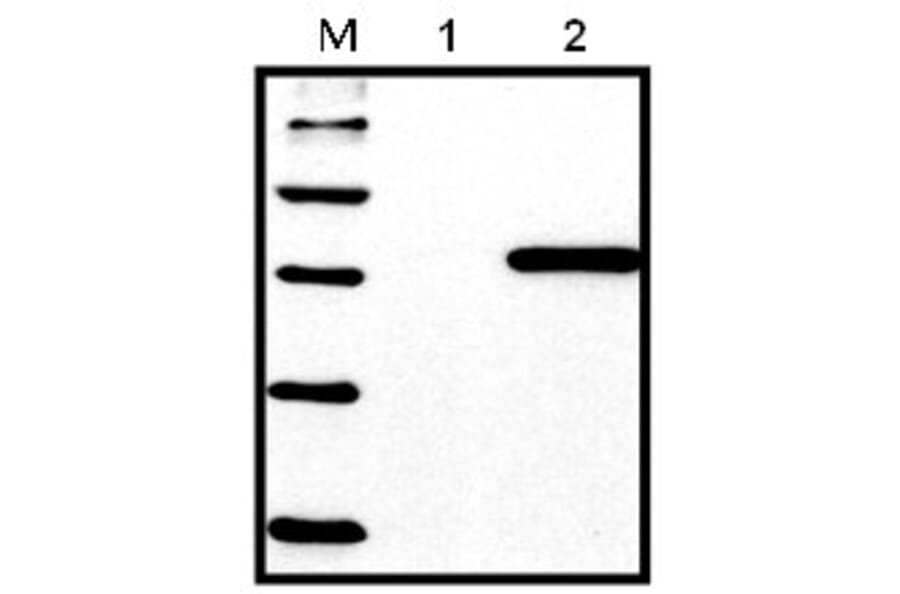
Western Blot - Anti-His Tag Antibody [HIS.H8] (A85277)
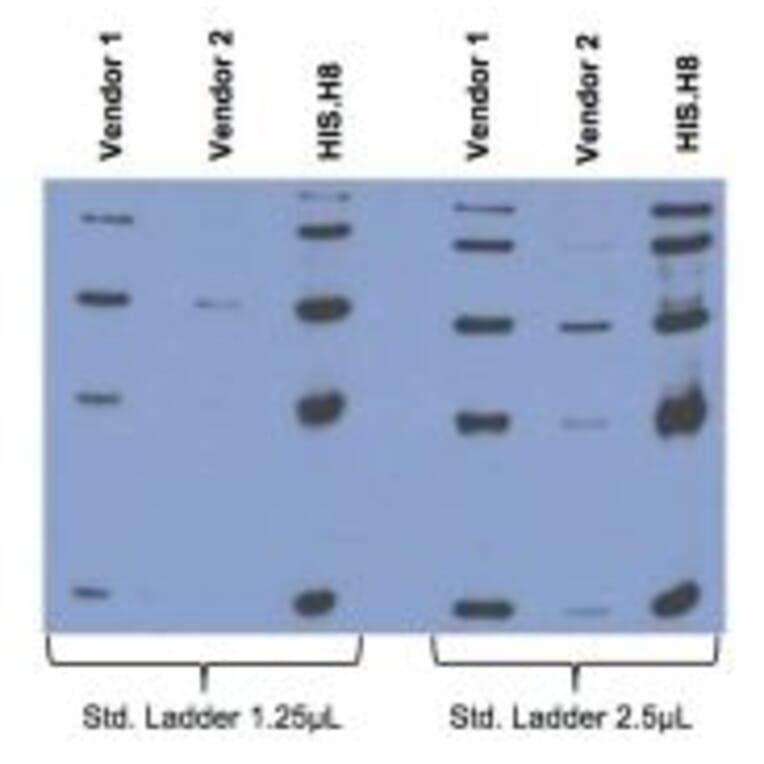
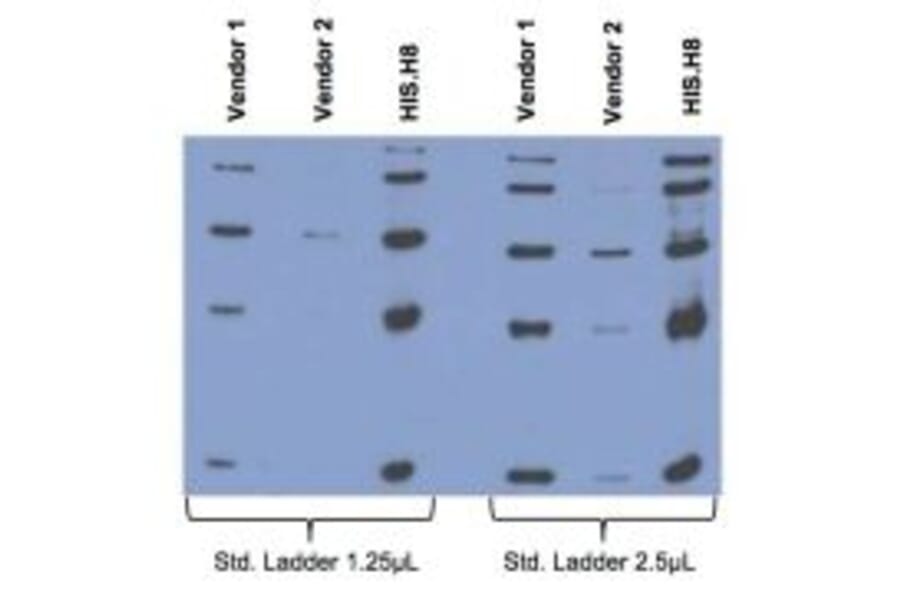
Comparison between Anti-His tag (HIS.H8 / EH158) mAb with 2 different vendor Abs, probed against a standard ladder containing five different His-tagged proteins. All Ab dilutions are 1:2000 (0.5µg/ml).
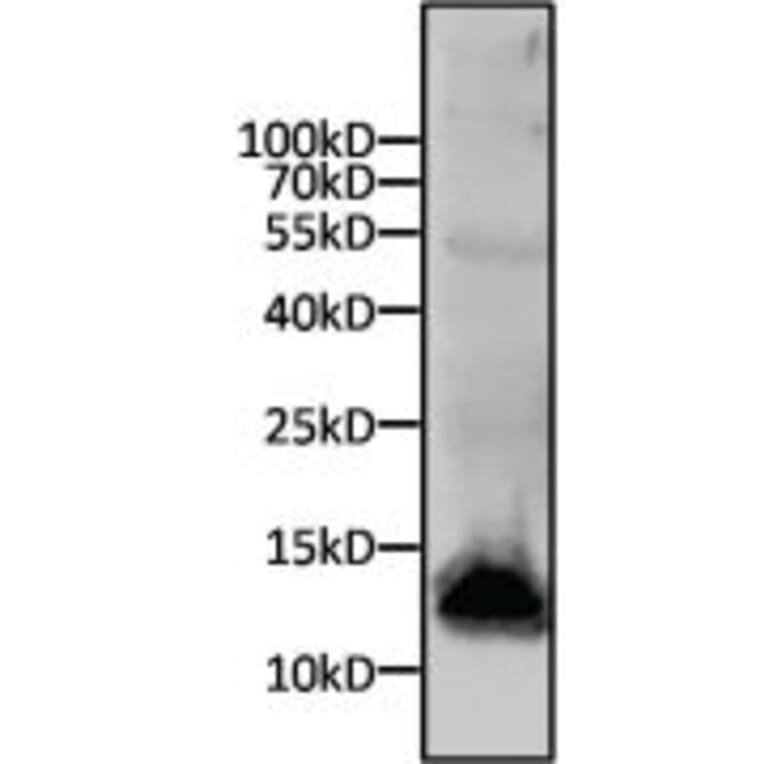
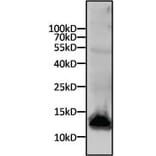
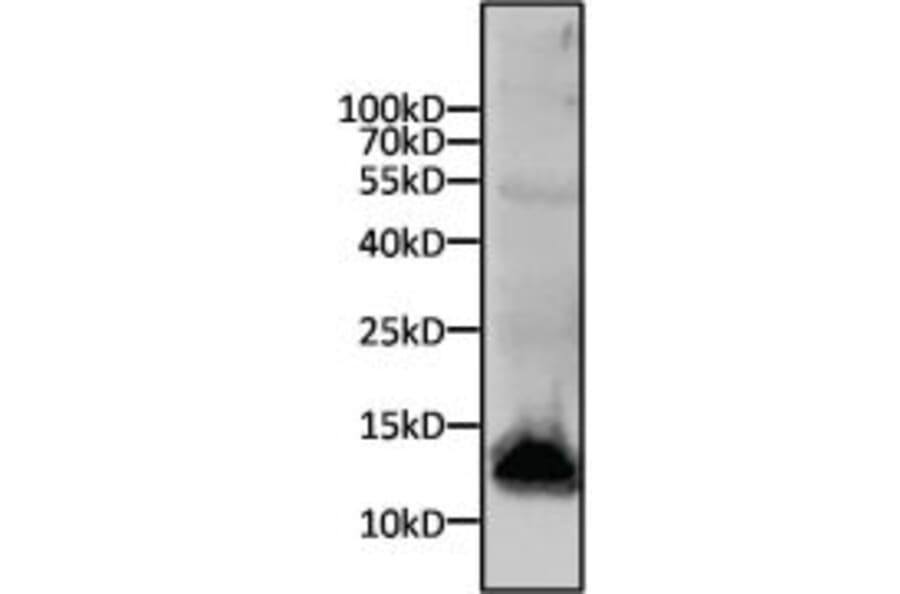
Western Blot - Anti-His Tag Antibody [HIS.H8] (A85277)
Western blot analysis of a 6x His-tagged protein was performed by loading 15µg of cell lysates from SF9 cells expressing an 11kD 6x-His tagged protein per well onto an SDS-PAGE gel. Proteins were transferred to a nitrocellulose membrane, and blocked in blocking buffer for 1 hour at room temperature. The membrane was probed with Anti-His Tag Antibody at a dilution of 1:500 overnight at 4 degrees celsius, washed in TBST, and probed with an Anti-Mouse IgG secondary antibody conjugated to a near-IR dye. Results were visualized using a near-IR imager.
Publishing research using Anti-His Tag Antibody [HIS.H8] (A85277)? Please let us know so that we can list the citation on this page.