Unconjugated
Here we demonstrate localization of the isoform3 of DNA Methyltransferase1 (DNMT1) enzyme to mitochondria, instead of isoform1 as reported earlier. The fused DNMT1-isoform1, reported earlier to localize in mitochondria, surprisingly showed its exclusive presence inside the nucleus after its ectopic expression; and failed to localize in mitochondria. On the other hand, ectopically expressed DNMT1-isoform3 targeted itself to mitochondria and subsequently methylated CpG regions in the mitochondrial genome. In addition, overexpression of DNMT1-isoform3 affected mitochondrial biology and regulated its function. Under different conditions of oxidative and nutritional stress, this isoform was down-regulated, resulting in hypomethylation of mitochondrial genome. Our study reveals how DNMT1-isoform3, instead of isoform1, is responsible for mtDNA methylation, influencing its biology.
Autophagy is an evolutionarily conserved cellular process that primarily participates in lysosome-mediated protein degradation. Although autophagy is a cytoplasmic event, how epigenetic pathways are involved in the regulation of autophagy remains incompletely understood. Here, we found that H2B monoubiquitination (H2Bub1) is down-regulated in cells under starvation conditions and that the decrease in H2Bub1 results in the activation of autophagy. We also identified that the deubiquitinase USP44 is responsible for the starvation-induced decrease in H2Bub1. Furthermore, the changes in H2Bub1 affect the transcription of genes involved in the regulation of autophagy. Therefore, this study reveals a novel epigenetic pathway for the regulation of autophagy through H2Bub1.
The dynamic interaction of DNA methylation and transcription factor binding in regulating spatiotemporal gene expression is essential for embryogenesis, but the underlying mechanisms remain understudied. In this study, using mouse models and integration of in vitro and in vivo genetic and epigenetic analyses, we show that the binding of REST (repressor element 1 (RE1) silencing transcription factor; also known as NRSF) to its cognate RE1 sequences is temporally regulated by non-CpG methylation. This process is dependent on DNA methyltransferase 3B (DNMT3B) and leads to suppression of adult cardiac genes in developing hearts. We demonstrate that DNMT3B preferentially mediates non-CpG methylation of REST-targeted genes in the developing heart. Downregulation of DNMT3B results in decreased non-CpG methylation of RE1 sequences, reduced REST occupancy, and consequently release of the transcription suppression during later cardiac development. Together, these findings reveal a critical gene silencing mechanism in developing mammalian hearts that is regulated by the dynamic interaction of DNMT3B-mediated non-CpG methylation and REST binding.
Autophagy is an evolutionarily conserved cellular process that primarily participates in lysosome-mediated protein degradation. Although autophagy is a cytoplasmic event, how epigenetic pathways are involved in the regulation of autophagy remains incompletely understood. Here, we found that H2B monoubiquitination (H2Bub1) is down-regulated in cells under starvation conditions and that the decrease in H2Bub1 results in the activation of autophagy. We also identified that the deubiquitinase USP44 is responsible for the starvation-induced decrease in H2Bub1. Furthermore, the changes in H2Bub1 affect the transcription of genes involved in the regulation of autophagy. Therefore, this study reveals a novel epigenetic pathway for the regulation of autophagy through H2Bub1.
DNA methylation is an important epigenetic mark that regulates gene expression. Dnmt1 plays an important role in maintaining DNA methylation patterns on daughter DNA strands. Studies have shed light into the functional role of Dnmt1 regulation in the hematopoietic and epidermal systems. Here we show that Dnmt1 is required for myogenesis. Loss of Dnmt1 results in reduced expression of myogenic genes and defects in myogenic differentiation. We have utilized a conditional knockout mouse approach to examine the functional consequences of Dnmt1 depletion specifically in the developing muscle. These mice were born runted, with smaller body weights, and reduced ability to form myotubes in vitro. We show that expression of Id-1, a negative regulator of myogenesis, is enhanced in Dnmt1-deficient cultures, leading to enhanced transdifferentiation of myoblasts toward the osteogenic lineage. Thus, these studies demonstrate that Dnmt1 influences cellular identity and determines lineage fidelity.
Tissue inhibitor of matrix metalloprotease 4 (TIMP4) is endogenously one of the key modulators of matrix metalloprotease 9 (MMP9) and we have reported earlier that cardiac specific TIMP4 instigates contractility and helps in differentiation of cardiac progenitor cells. Although studies show that the expression of TIMP4 goes down in heart failure but the mechanism is unknown. This study aims to determine the mechanism of silencing of TIMP4 in heart failure progression created by aorta-vena cava (AV) fistula. We hypothesize that there is epigenetic silencing of TIMP4 in heart failure. To validate this hypothesis, we created heart failure model by creating AV fistula in C57BL/6 mice and looked into the promoter methylation (methylation specific PCR, high resolution melting, methylation sensitive restriction enzyme and Na bisulphite treatment followed by sequencing), histone modification (ChIP assay) and microRNAs that regulate TIMP4 (mir122a) and MMP9 (mir29b and mir455-5p). The physiological parameters in terms of cardiac function after AV fistula were assessed by echocardiography. We observed that there are 7 CpG islands in the TIMP4 promoter which get methylated during the progression of heart failure which leads to its epigenetic silencing. In addition, the up-regulated levels of mir122a in part, contribute to regulation of TIMP4. Consequently, MMP9 gets up-regulated and leads to cardiac remodeling. This is a novel report to explain the epigenetic silencing of TIMP4 in heart failure.
© 2016 The Authors. Journal of Cellular and Molecular Medicine published by John Wiley & Sons Ltd and Foundation for Cellular and Molecular Medicine.
A common single-nucleotide polymorphism in the telomerase reverse transcriptase (TERT) promoter, rs2853669 influences patient survival rates and the risk of developing cancer. Recently, several lines of evidence suggest that the rs2853669 suppresses TERT promoter mutation-mediated TERT expression levels and cancer mortality as well as recurrence rates. However, no reports are available on the impact of rs2853669 on TERT expression in hepatocellular carcinoma (HCC) and its association with patient survival. Here, we found that HCC-related overall and recurrence-free survival rates were not associated with TERT promoter mutation individually, but rs2853669 and the TERT promoter mutation in combination were associated with poor survival rates. TERT mRNA expression and telomere fluorescence levels were greater in patients with HCC who had both the combination. The combination caused TERT promoter methylation through regulating the binding of DNA methyltransferase 1 and histone deacetylase 1 to the TERT promoter in HCC cell lines. The TERT expression level was significantly higher in HCC tumor with a methylated promoter than in that with an unmethylated promoter. In conclusion, we demonstrate a substantial role for the rs2853669 in HCC with TERT promoter mutation, which suggests that the combination of the rs2853669 and the mutation indicate poor prognoses in liver cancer.
Epigenetic silencing including histone modifications and DNA methylation is an important tumorigenic mechanism. However, its role in cancer immunopathology and immunotherapy is poorly understood. Using human ovarian cancers as our model, here we show that enhancer of zeste homologue 2 (EZH2)-mediated histone H3 lysine 27 trimethylation (H3K27me3) and DNA methyltransferase 1 (DNMT1)-mediated DNA methylation repress the tumour production of T helper 1 (TH1)-type chemokines CXCL9 and CXCL10, and subsequently determine effector T-cell trafficking to the tumour microenvironment. Treatment with epigenetic modulators removes the repression and increases effector T-cell tumour infiltration, slows down tumour progression, and improves the therapeutic efficacy of programmed death-ligand 1 (PD-L1; also known as B7-H1) checkpoint blockade and adoptive T-cell transfusion in tumour-bearing mice. Moreover, tumour EZH2 and DNMT1 are negatively associated with tumour-infiltrating CD8(+) T cells and patient outcome. Thus, epigenetic silencing of TH1-type chemokines is a novel immune-evasion mechanism of tumours. Selective epigenetic reprogramming alters the T-cell landscape in cancer and may enhance the clinical efficacy of cancer therapy.
Human papillomavirus (HPV) oncoproteins drive distinctive promoter methylation patterns in cancer. However, the underlying mechanism remains to be elucidated. Cyclin A1 (CCNA1) promoter methylation is strongly associated with HPV-associated cancer. CCNA1 methylation is found in HPV-associated cervical cancers, as well as in head and neck squamous cell cancer. Numerous pieces of evidence suggest that E7 may drive CCNA1 methylation. First, the CCNA1 promoter is methylated in HPV-positive epithelial lesions after transformation. Second, the CCNA1 promoter is methylated at a high level when HPV is integrated into the human genome. Finally, E7 has been shown to interact with DNA methyltransferase 1 (Dnmt1). Here, we sought to determine the mechanism by which E7 increases methylation in cervical cancer by using CCNA1 as a gene model. We investigated whether E7 induces CCNA1 promoter methylation, resulting in the loss of expression. Using both E7 knockdown and overexpression approaches in SiHa and C33a cells, our data showed that CCNA1 promoter methylation decreases with a corresponding increase in expression in E7 siRNA-transfected cells. By contrast, CCNA1 promoter methylation was augmented with a corresponding reduction in expression in E7-overexpressing cells. To confirm whether the binding of the E7-Dnmt1 complex to the CCNA1 promoter induced methylation and loss of expression, ChIP assays were carried out in E7-, del CR3-E7 and vector control-overexpressing C33a cells. The data showed that E7 induced CCNA1 methylation by forming a complex with Dnmt1 at the CCNA1 promoter, resulting in the subsequent reduction of expression in cancers. It is interesting to further explore the genome-wide mechanism of E7 oncoprotein-mediated DNA methylation.
Adiponectin plays a key role in the regulation of the whole-body energy homeostasis by modulating glucose and lipid metabolism. Although obesity-induced reduction of adiponectin expression is primarily ascribed to a transcriptional regulation failure, the underlying mechanisms are largely undefined. Here we show that DNA hypermethylation of a particular region of the adiponectin promoter suppresses adiponectin expression through epigenetic control and, in turn, exacerbates metabolic diseases in obesity. Obesity-induced, pro-inflammatory cytokines promote DNMT1 expression and its enzymatic activity. Activated DNMT1 selectively methylates and stimulates compact chromatin structure in the adiponectin promoter, impeding adiponectin expression. Suppressing DNMT1 activity with a DNMT inhibitor resulted in the amelioration of obesity-induced glucose intolerance and insulin resistance in an adiponectin-dependent manner. These findings suggest a critical role of adiponectin gene epigenetic control by DNMT1 in governing energy homeostasis, implying that modulating DNMT1 activity represents a new strategy for the treatment of obesity-related diseases.
Krüppel-like factor 4 (KLF4) is a transcription factor which plays divergent roles in a number of physiological or pathological process. However, the expression and role of KLF4 in renal fibrosis remain undetermined. The aim of the present study was to determine the epigenetic alterations of KLF4 and its potential role and mechanisms of action in epithelial-to-mesenchymal transition (EMT) in renal fibrosis. The hypermethylation of the KLF4 promoter accompanied by a decrease in KLF4 expression were observed in mice subjected to unilateral ureteral obstruction (UUO) and in HK-2 cells stimulated with transforming growth factor (TGF)-ß1. However, treatment with 5-aza-2'-deoxycytidine attenuated the TGF-ß1-induced downregulation of KLF4 and E-cadherin and the upregulation of a-smooth muscle actin (a-SMA) in the HK-2 cells. DNA methyltransferase 1 (Dnmt1) participated in the TGF-ß1-mediated hypermethylation of the KLF4 promoter in the HK-2 cells. In addition, functional analysis demonstrated that the overexpression of KLF4 led to an increase in the expression of E-cadherin and zonula occludens-l (ZO-1), and a decrease in the expression of a-SMA and fibroblast-specific protein 1 (FSP-1), thus reversing the effects of the suppression of KLF4. These data suggest that KLF4 inhibits the progression of EMT in renal epithelial cells. In conclusion, our findings demonstrate that KLF4 is downregulated during EMT in renal fibrosis in vivo and in vitro; thus, KLF4 functions as a suppressor of renal fibrogenesis. The hypermethylation of KLF4 directly mediated by Dnmt1 contributes to the progression of EMT in renal epithelial cells. KLF4 promoter methylation may thus be a promising diagnostic marker or therapeutic target in renal fibrosis.
X-chromosome inactivation (XCI), the random transcriptional silencing of one X chromosome in somatic cells of female mammals, is a mechanism that ensures equal expression of X-linked genes in both sexes. XCI is initiated in cis by the noncoding Xist RNA, which coats the inactive X chromosome (Xi) from which it is produced. However, trans-acting factors that mediate XCI remain largely unknown. Here, we perform a large-scale RNA interference screen to identify trans-acting XCI factors (XCIFs) that comprise regulators of cell signaling and transcription, including the DNA methyltransferase, DNMT1. The expression pattern of the XCIFs explains the selective onset of XCI following differentiation. The XCIFs function, at least in part, by promoting expression and/or localization of Xist to the Xi. Surprisingly, we find that DNMT1, which is generally a transcriptional repressor, is an activator of Xist transcription. Small-molecule inhibitors of two of the XCIFs can reversibly reactivate the Xi, which has implications for treatment of Rett syndrome and other dominant X-linked diseases. A homozygous mouse knockout of one of the XCIFs, stanniocalcin 1 (STC1), has an expected XCI defect but surprisingly is phenotypically normal. Remarkably, X-linked genes are not overexpressed in female Stc1(-/-) mice, revealing the existence of a mechanism(s) that can compensate for a persistent XCI deficiency to regulate X-linked gene expression.
We report evidence of a detailed epigenetic modification of the leptin promoter and the effects of n-3 polyunsaturated fatty acids (n-3 PUFAs), which is closely associated with the leptin gene transcription in obesity. In the adipose tissue of diet induced obese (DIO) mice, methylation of the CpG island and the binding of methyl-CpG-binding domain protein 2 (MBD2) and DNA methyltransferases (DNMTs) at the leptin promoter are increased and RNA Pol II is decreased. Additionally, histones H3 and H4 are hypoacetylated, lysine 4 of histone H3 (H3K4) is hypomethylated and the binding of histone deacetylases (HDACs) 1, 2 and 6 is increased at the leptin promoter in the DIO mice. These modifications may serve a feedback role to maintain leptin concentrations within a normal range. The regulation of leptin transcriptional expression by n-3 PUFAs is mediated, at least in part, by epigenetic targets, such as MBD2 and histone modifications.
Epigenetic gene silencing by histone modifications and DNA methylation is essential for cancer development. The molecular mechanism that promotes selective epigenetic changes during tumorigenesis is not understood. We report here that the PIAS1 SUMO ligase is involved in the progression of breast tumorigenesis. Elevated PIAS1 expression was observed in breast tumor samples. PIAS1 knockdown in breast cancer cells reduced the subpopulation of tumor-initiating cells, and inhibited breast tumor growth in vivo. PIAS1 acts by delineating histone modifications and DNA methylation to silence the expression of a subset of clinically relevant genes, including breast cancer DNA methylation signature genes such as cyclin D2 and estrogen receptor, and breast tumor suppressor WNT5A. Our studies identify a novel epigenetic mechanism that regulates breast tumorigenesis through selective gene silencing.
RGS10 is an important regulator of cell survival and chemoresistance in ovarian cancer. We recently showed that RGS10 transcript expression is suppressed during acquired chemoresistance in ovarian cancer. The suppression of RGS10 is due to DNA hypermethylation and histone deacetylation, two important mechanisms that contribute to silencing of tumor suppressor genes during cancer progression. Here, we fully investigate the molecular mechanisms of epigenetic silencing of RGS10 expression in chemoresistant A2780-AD ovarian cancer cells. We identify two important epigenetic regulators, HDAC1 and DNMT1, that exhibit aberrant association with RGS10 promoters in chemoresistant ovarian cancer cells. Knockdown of HDAC1 or DNMT1 expression, and pharmacological inhibition of DNMT or HDAC enzymatic activity, significantly increases RGS10 expression and cisplatin-mediated cell death. Finally, DNMT1 knock down also decreases HDAC1 binding to the RGS10 promoter in chemoresistant cells, suggesting HDAC1 recruitment to RGS10 promoters requires DNMT1 activity. Our results suggest that HDAC1 and DNMT1 contribute to the suppression of RGS10 during acquired chemoresistance and support inhibition of HDAC1 and DNMT1 as an adjuvant therapeutic approach to overcome ovarian cancer chemoresistance.
Methyltransferase expression and DNA methylation are linked to aging and age-related disease. We utilized 3-, 12-, and 24-month-old Ames dwarf and their wild-type siblings to examine the genotype and age-related differences in the expression of methyltransferase enzymes related to DNA methylation in the liver, glycine-N-methyltransferase and DNA methyltransferase (DNMT). We found that DNMT proteins and transcripts are differentially expressed in dwarf mice compared with wild-type siblings that can be attributed to age and/or genotype. However, DNMT1 protein expression is drastically reduced compared with wild-type controls at every age. DNMT3a protein levels coincide with differences observed in DNMT activity. Growth hormone appears to modulate expression of DNMT1 and 3a in dwarf liver tissue and primary hepatocytes. Therefore, growth hormone may contribute to age-related processes, DNA methylation, and, ultimately, longevity.
Deterioration of the immune system (immunosenescence) with age is associated with an increased susceptibility to infection, autoimmune disease and cancer, and reduced responsiveness to vaccination. Immunosenescence entails a reduced supply of naïve T cells from the thymus and increased specialization of peripheral T cell clones. Both thymic involution and peripheral T cell homeostasis are thought to involve cellular senescence. In order to analyze this at the molecular level, we studied gene expression profiles, epigenetic status, and genome stability in the thymus and spleen of 1-, 4-, and 18-month-old Long Evans rats. In the thymus, altered gene expression, DNA and histone H3K9 hypomethylation, increased genome instability, and apoptosis were observed in 18-month-old animals compared to 1- and 4-month-old animals. In the spleen, alterations in gene expression and epigenetic regulation occurred already by the age of 4 months compared to 1 month and persisted in 18-month-old compared to 1-month-old rats. In both organs, these changes were accompanied by the altered composition of resident T cell populations. Our study suggests that both senescence and apoptosis may be involved in altered organ function.
We report that homology-directed repair of a DNA double-strand break within a single copy Green Fluorescent Protein (GFP) gene in HeLa cells alters the methylation pattern at the site of recombination. DNA methyl transferase (DNMT)1, DNMT3a and two proteins that regulate methylation, Np95 and GADD45A, are recruited to the site of repair and are responsible for selective methylation of the promoter-distal segment of the repaired DNA. The initial methylation pattern of the locus is modified in a transcription-dependent fashion during the 15-20 days following repair, at which time no further changes in the methylation pattern occur. The variation in DNA modification generates stable clones with wide ranges of GFP expression. Collectively, our data indicate that somatic DNA methylation follows homologous repair and is subjected to remodeling by local transcription in a discrete time window during and after the damage. We propose that DNA methylation of repaired genes represents a DNA damage code and is source of variation of gene expression.
DNA methylation was first described almost a century ago; however, the rules governing its establishment and maintenance remain elusive. Here we present data demonstrating that active transcription regulates levels of genomic methylation. We identify a novel RNA arising from the CEBPA gene locus that is critical in regulating the local DNA methylation profile. This RNA binds to DNMT1 and prevents CEBPA gene locus methylation. Deep sequencing of transcripts associated with DNMT1 combined with genome-scale methylation and expression profiling extend the generality of this finding to numerous gene loci. Collectively, these results delineate the nature of DNMT1-RNA interactions and suggest strategies for gene-selective demethylation of therapeutic targets in human diseases.
The retinoblastoma tumor suppressor gene (RB) product has been implicated in epigenetic control of gene expression owing to its ability to physically bind to many chromatin modifiers. However, the biological and clinical significance of this activity was not well elucidated. To address this, we performed genetic and epigenetic analyses in an Rb-deficient mouse thyroid C cell tumor model. Here we report that the genetic interaction of Rb and ATM regulates DNMT1 protein stability and hence controls the DNA methylation status in the promoters of at least the Ink4a, Shc2, FoxO6, and Noggin genes. Furthermore, we demonstrate that inactivation of pRB promotes Tip60 (acetyltransferase)-dependent ATM activation; allows activated ATM to physically bind to DNMT1, forming a complex with Tip60 and UHRF1 (E3 ligase); and consequently accelerates DNMT1 ubiquitination driven by Tip60-dependent acetylation. Our results indicate that inactivation of the pRB pathway in coordination with aberration in the DNA damage response deregulates DNMT1 stability, leading to an abnormal DNA methylation pattern and malignant progression.
A number of proteins form covalent bonds with DNA as obligatory transient intermediates in normal nuclear transactions. Drugs that trap these complexes have proven to be potent therapeutics in both cancer and infectious disease. Nonetheless, current assays for DNA-protein adducts are cumbersome, limiting both mechanistic studies and translational applications. We have developed a rapid and sensitive assay that enables quantitative immunodetection of protein-DNA adducts. This new 'RADAR' (rapid approach to DNA adduct recovery) assay accelerates processing time 4-fold, increases sample throughput 20-fold and requires 50-fold less starting material than the current standard. It can be used to detect topoisomerase 1-DNA adducts in as little as 60 ng of DNA, corresponding to 10 000 human cells. We apply the RADAR assay to demonstrate that expression of SLFN11 does not increase camptothecin sensitivity by promoting accumulation of topoisomerase 1-DNA adducts. The RADAR assay will be useful for analysis of the mechanisms of formation and resolution of DNA-protein adducts in living cells, and identification and characterization of reactions in which covalent DNA adducts are transient intermediates. The assay also has potential application to drug discovery and individualized medicine.
We tested the hypothesis that miR-133a regulates DNA methylation by inhibiting Dnmt-1 (maintenance) and Dnmt-3a and -3b (de novo) methyl transferases in diabetic hearts by using Ins2(+/-) Akita (diabetic) and C57BL/6J (WT), mice and HL1 cardiomyocytes. The specific role of miR-133a in DNA methylation in diabetes was assessed by two treatment groups (1) scrambled, miR-133a mimic, anti-miR-133a, and (2) 5mM glucose (CT), 25 mM glucose (HG) and HG+miR-133a mimic. The levels of miR-133a, Dnmt-1, -3a and -3b were measured by multiplex RT-PCR, qPCR and Western blotting. The results revealed that miR-133a is inhibited but Dnmt-1 and -3b are induced in Akita suggesting that attenuation of miR-133a induces both maintenance (Dnmt-1) - and de novo - methylation (Dnmt-3b) in diabetes. The up regulation of Dnmt-3a in Akita hearts elicits intricate and antagonizing interaction between Dnmt-3a and -3b. In cardiomyocytes, over expression of miR-133a inhibits but silencing of miR-133a induces Dnmt-1, -3a and -3b elucidating the involvement of miR-133a in regulation of DNA methylation. The HG treatment up regulates only Dnmt-1 and not Dnmt-3a and -3b suggesting that acute hyperglycemia triggers only maintenance methylation. The over expression of miR-133a mitigates glucose mediated induction of Dnmt-1 illustrating the role of miR-133a in regulation of DNA methylation in diabetes.
Methylation of CpG islands inactivates transcription of tumor suppressor genes including p16 (CDKN2A). Inhibitors of DNA methylation and histone deacylation are recognized as useful cancer therapeutic chemicals through reactivation of the expression of methylated genes. However, these inhibitors are not target gene-specific, so that they lead to serious side effects as regular cytotoxic chemotherapy agents. To explore the feasibility of methylated gene-specific reactivation by artificial transcription factors, we engineered a set of Sp1-like seven-finger zinc-finger proteins (7ZFPs) targeted to a 21-bp sequence of the p16 promoter and found that these 7ZFPs could bind specifically to the target p16 promoter probe. Then the p16-specific artificial transcription factors (p16ATFs) were made from these 7ZFPs and the transcription activator VP64. Results showed that transient transfection of some p16ATFs selectively up-regulated the endogenous p16 expression in the p16-active 293T cells. Moreover, the transient transfection of the representative p16ATF-6I specifically reactivated p16 expression in the p16-methylated H1299 and AGS cells pretreated with a nontoxic amount of 5'-aza-deoxycytidine (20 and 80 nM, respectively). In addition, stable transfection of the p16ATF induced demethylation of p16 CpG island and trimethylation of histone H3K4, and inhibited recruitment of DNA methyltransferase 1 and trimethylation of H3K9 and H3K27 in the p16 promoter in H1299 cells without 5'-aza-deoxycytidine pretreatment. Notably, inhibition of cell migration and invasion was observed in these p16-reactivated cells induced by transient and stable p16ATF transfection. These results demonstrate that p16ATF not only specifically reactivates p16 expression through demethylation of CpG islands, but also restores methylated p16 function.
DNA methylation is an epigenetic mechanism establishing long-term gene silencing during development and cell commitment, which is maintained in subsequent cell generations. Aberrant DNA methylation is found at gene promoters in most cancers and can lead to silencing of tumor suppressor genes. The DNA methyltransferase inhibitor 5-aza-2'-deoxycytidine (5-aza-CdR) is able to reactivate genes silenced by DNA methylation and has been shown to be a very potent epigenetic drug in several hematological malignancies. In this report, we demonstrate that 5-aza-CdR exhibits high antineoplastic activity against anaplastic large cell lymphoma (ALCL), a rare CD30 positive non-Hodgkin lymphoma of T-cell origin. Low dose treatment of ALCL cell lines and xenografted tumors causes apoptosis and cell cycle arrest in vitro and in vivo. This is also reflected in genome-wide expression analyses, where genes related to apoptosis and cell death are amongst the most affected targets of 5-aza-CdR. Furthermore, we observed demethylation and re-expression of p16(INK4A) after drug administration and senescence associated ß-galactosidase activity. Thus, our data provide evidence that 5-aza-CdR is highly efficient against ALCL and warrants further clinical evaluation for future therapeutic use.
Zebularine is a novel potent inhibitor of both cytidine deaminase and DNA methylation. We examined the effect of zebularine on mammary tumor growth in genetically engineered MMTV-PyMT transgenic mice that develop mammary tumors at 60 days of age with 100% penetrance. The MMTV-PyMT transgenic mice were randomized at 46 days of age into control (n = 25) and zebularine (n = 25) treatment groups and monitored for parameters of tumor growth. Zebularine was administered at 5 mg/mL in drinking water. We observed a significant delay in the growth of mammary tumors in zebularine-treated mice with a statistically significant reduction (P = 0.0135) in total tumor burden at 94 days of age when the mice were sacrificed. After 48 days of zebularine treatment, the tumors were predominantly necrotic compared with untreated animals. In addition, a high apoptotic index by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling assay was observed as early as 13 days following treatment. Immunoblot analysis showed depletion of DNMT1 and partial depletion of DNMT3b after zebularine treatment. Microarray analyses of global gene expression identified upregulation of twelve methylation-regulated genes as well as a set of candidate cancer genes that participate in cell growth and apoptosis. In summary, zebularine inhibits the growth of spontaneous mammary tumors and causes early onset of tumor cell necrosis and apoptosis in a genetically engineered mouse model of breast cancer. Defining the parameters of zebularine-mediated tumor inhibition may advance the future development of DNA methyltransferase inhibitors as an effective cancer treatment.
Apoptosis genes, such as TP53 and p16/CDKN2A, that mediate responses to cytotoxic chemotherapy, are frequently nonfunctional in melanoma. Differentiation may be an alternative to apoptosis for inducing melanoma cell cycle exit. Epigenetic mechanisms regulate differentiation, and DNA methylation alterations are associated with the abnormal differentiation of melanoma cells. The effects of the deoxycytidine analogue decitabine (5-aza-2'-deoxycytidine), which depletes DNA methyl transferase 1 (DNMT1), on melanoma differentiation were examined. Treatment of human and murine melanoma cells in vitro with concentrations of decitabine that did not cause apoptosis inhibited proliferation accompanied by cellular differentiation. A decrease in promoter methylation, and increase in expression of the melanocyte late-differentiation driver SOX9, was followed by increases in cyclin-dependent kinase inhibitors (CDKN) p27/CDKN1B and p21/CDKN1A that mediate cell cycle exit with differentiation. Effects were independent of the TP53, p16/CDKN2A and also the BRAF status of the melanoma cells. Resistance, when observed, was pharmacologic, characterized by diminished ability of decitabine to deplete DNMT1. Treatment of murine melanoma models in vivo with intermittent, low-dose decitabine, administered sub-cutaneously to limit high peak drug levels that cause cytotoxicity and increase exposure time for DNMT1 depletion, and with tetrahydrouridine to decrease decitabine metabolism and further increase exposure time, inhibited tumor growth and increased molecular and tumor stromal factors implicated in melanocyte differentiation. Modification of decitabine dose, schedule and formulation for differentiation rather than cytotoxic objectives inhibits the growth of melanoma cells in vitro and in vivo.
Although Epstein-Barr virus (EBV) usually establishes an asymptomatic lifelong infection, it is also implicated in the development of germinal center (GC) B-cell-derived malignancies, including Hodgkin's lymphoma (HL). Following primary infection, EBV remains latent in the memory B-cell population, where host-driven methylation of viral DNA contributes to the repression of viral gene expression. However, it is still unclear how EBV harnesses the cell's methylation machinery in B cells, how this contributes to viral persistence, and what impact this has on the methylation of cellular genes. We show that EBV infection of GC B cells is followed by upregulation of the DNA methyltransferase DNMT3A and downregulation of DNMT3B and DNMT1. We show that the EBV latent membrane protein 1 (LMP1) oncogene downregulates DNMT1 and that DNMT3A binds to the viral promoter Wp. Genome-wide promoter arrays performed with these cells showed that EBV-associated methylation changes in cellular genes were not randomly distributed across the genome but clustered at chromosomal locations, consistent with an instructive pattern of methylation, and were in part determined by promoter CpG content. Both DNMT3B and DNMT1 were downregulated and DNMT3A was upregulated in HL cell lines, recapitulating the pattern of expression observed following EBV infection of GC B cells. We also found, by using gene expression profiling, that genes differentially expressed following EBV infection of GC B cells were significantly enriched for those reported to be differentially expressed in HL. These observations suggest that EBV-infected GC B cells are a useful model for studying virus-associated changes contributing to the pathogenesis of HL.
High levels of DNA methyltransferase 1 (DNMT1), hypermethylation, and downregulation of GAD(67) and reelin have been described in GABAergic interneurons of patients with schizophrenia (SZ) and bipolar (BP) disorders. However, overexpression of DNMT1 is lethal, making it difficult to assess the direct effect of high levels of DNMT1 on neuronal development in vivo. We therefore used Dnmt1(tet/tet) mouse ES cells that overexpress DNMT1 as an in vitro model to investigate the impact of high levels of DNMT1 on neuronal differentiation. Although there is down-regulation of DNMT1 during early stages of differentiation in wild type and Dnmt1(tet/tet) ES cell lines, neurons derived from Dnmt1(tet/tet) cells showed abnormal dendritic arborization and branching. The Dnmt1(tet/tet) neuronal cells also showed elevated levels of functional N-methyl d-aspartate receptor (NMDAR), a feature also reported in some neurological and neurodegenerative disorders. Considering the roles of reelin and GAD(67) in neuronal networking and excitatory/inhibitory balance, respectively, we studied methylation of these genes' promoters in Dnmt1(tet/tet) ES cells and neurons. Both reelin and GAD(67) promoters were not hypermethylated in the Dnmt1(tet/tet) ES cells and neurons, suggesting that overexpression of DNMT1 may not directly result in methylation-mediated repression of these two genes. Taken together, our results suggest that overexpression of DNMT1 in ES cells results in an epigenetic change prior to the onset of differentiation. This epigenetic change in turn results in abnormal neuronal differentiation and upregulation of functional NMDA receptor.
Current drug therapy for metastatic renal cell cancer (RCC) results in temporary disease control but not cure, necessitating continued investigation into alternative mechanistic approaches. Drugs that inhibit chromatin-modifying enzymes involved in transcription repression (chromatin-relaxing drugs) could have a role, by inducing apoptosis and/or through differentiation pathways. At low doses, the cytosine analogue decitabine (DAC) can be used to deplete DNA methyl-transferase 1 (DNMT1), modify chromatin, and alter differentiation without causing apoptosis (cytotoxicity). Noncytotoxic regimens of DAC were evaluated for in vitro and in vivo efficacy against RCC cell lines, including a p53-mutated RCC cell line developed from a patient with treatment-refractory metastatic RCC. The cell division-permissive mechanism of action-absence of early apoptosis or DNA damage, increase in expression of HNF4a (hepatocyte nuclear factor 4a), a key driver associated with the mesenchymal to epithelial transition, decrease in mesenchymal marker expression, increase in epithelial marker expression, and late increase in cyclin-dependent kinase inhibitor CDKN1B (p27) protein-was consistent with differentiation-mediated cell-cycle exit. In vivo blood counts and animal weights were consistent with minimal toxicity of therapy. The distinctive mechanism of action of a dose and schedule of DAC designed for noncytotoxic depletion of DNMT1 suggests a potential role in treating RCC.
Large fractions of eukaryotic genomes contain repetitive sequences of which the vast majority is derived from transposable elements (TEs). In order to inactivate those potentially harmful elements, host organisms silence TEs via methylation of transposon DNA and packaging into chromatin associated with repressive histone marks. The contribution of individual histone modifications in this process is not completely resolved. Therefore, we aimed to define the role of reversible histone acetylation, a modification commonly associated with transcriptional activity, in transcriptional regulation of murine TEs. We surveyed histone acetylation patterns and expression levels of ten different murine TEs in mouse fibroblasts with altered histone acetylation levels, which was achieved via chemical HDAC inhibition with trichostatin A (TSA), or genetic inactivation of the major deacetylase HDAC1. We found that one LTR retrotransposon family encompassing virus-like 30S elements (VL30) showed significant histone H3 hyperacetylation and strong transcriptional activation in response to TSA treatment. Analysis of VL30 transcripts revealed that increased VL30 transcription is due to enhanced expression of a limited number of genomic elements, with one locus being particularly responsive to HDAC inhibition. Importantly, transcriptional induction of VL30 was entirely dependent on the activation of MAP kinase pathways, resulting in serine 10 phosphorylation at histone H3. Stimulation of MAP kinase cascades together with HDAC inhibition led to simultaneous phosphorylation and acetylation (phosphoacetylation) of histone H3 at the VL30 regulatory region. The presence of the phosphoacetylation mark at VL30 LTRs was linked with full transcriptional activation of the mobile element. Our data indicate that the activity of different TEs is controlled by distinct chromatin modifications. We show that activation of a specific mobile element is linked to a dual epigenetic mark and propose a model whereby phosphoacetylation of histone H3 is crucial for full transcriptional activation of VL30 elements.
The involvement of epigenetic alterations in mediating effects of estrogens on memory is unknown. The present study determined whether histone acetylation and DNA methylation are critical for the potent estrogen 17beta-estradiol (E(2)) to enhance object recognition memory. We show that dorsal hippocampal E(2) infusion increases acetylation of dorsal hippocampal histone H3, but not H4--an effect blocked by dorsal hippocampal inhibition of ERK activation. Further, intrahippocampal inhibition of ERK activation or DNA methyltransferase (DNMT) activity blocked the memory-enhancing effects of E(2). Consistent with these effects, E(2) decreased levels of HDAC2 protein and increased DNMT expression in the dorsal hippocampus. These findings provide evidence that the beneficial effects of E(2) on memory consolidation are associated with epigenetic alterations, and suggest these can be triggered by dorsal hippocampal ERK signaling.
The polycomb repressive complex (PRC) 2 contains 3 core proteins, EZH2, SUZ12, and EED, in which the SET (suppressor of variegation-enhancer of zeste-trithorax) domain of EZH2 mediates the histone methyltransferase activity. This induces trimethylation of lysine 27 on histone H3, regulates the expression of HOX genes, and promotes proliferation and aggressiveness of neoplastic cells. In this study, we demonstrate that treatment with the S-adenosylhomocysteine hydrolase inhibitor 3-deazaneplanocin A (DZNep) depletes EZH2 levels, and inhibits trimethylation of lysine 27 on histone H3 in the cultured human acute myeloid leukemia (AML) HL-60 and OCI-AML3 cells and in primary AML cells. DZNep treatment induced p16, p21, p27, and FBXO32 while depleting cyclin E and HOXA9 levels. Similar findings were observed after treatment with small interfering RNA to EZH2. In addition, DZNep treatment induced apoptosis in cultured and primary AML cells. Furthermore, compared with treatment with each agent alone, cotreatment with DZNep and the pan-histone deacetylase inhibitor panobinostat caused more depletion of EZH2, induced more apoptosis of AML, but not normal CD34(+) bone marrow progenitor cells, and significantly improved survival of nonobese diabetic/severe combined immunodeficiency mice with HL-60 leukemia. These findings indicate that the combination of DZNep and panobinostat is effective and relatively selective epigenetic therapy against AML cells.
5-aza-2'-deoxycytidine (DAC) is approved for the treatment of myelodysplastic syndromes, but resistance to this agent is common. In search for mechanisms of resistance, we measured the half maximal (50%) inhibitory concentration (IC(50)) of DAC and found it differed 1000-fold among a panel of cancer cell lines. The IC(50) was correlated with the doses of DAC that induced the most hypomethylation of long interspersed nuclear elements (LINE; R = 0.94, P < .001), but not with LINE methylation or DNA methyltransferase 1 (DNMT1), 3a, and 3b expression at baseline. Sensitivity to DAC showed a low correlation (R = 0.44, P = .11) to that of 5-azacytidine (AZA), but a good correlation to that of cytarabine (Ara-C; R = 0.89, P < .001). The 5 cell lines most resistant to DAC had a combination of low dCK, hENT1, and 2 transporters, and high cytosine deaminase. In an HL60 clone, resistance to DAC could be rapidly induced by drug exposure and was related to a switch from heterozygous to homozygous mutation of DCK. Transfection of wild-type DCK restored DAC sensitivity. DAC induced DNA breaks as evidenced by H2AX phosphorylation and increased homologous recombination rates by 7- to 10-fold. These results suggest that in vitro resistance to DAC can be explained by insufficient incorporation into DNA.
Histone deacetylase inhibitor (HDACi) has been shown to demethylate the mammalian genome, which further strengthens the concept that DNA methylation and histone modifications interact in regulation of gene expression. Here, we report that an HDAC inhibitor, depsipeptide, exhibited significant demethylating activity on the promoters of several genes, including p16, SALL3, and GATA4 in human lung cancer cell lines H719 and H23, colon cancer cell line HT-29, and pancreatic cancer cell line PANC1. Although expression of DNA methyltransferase 1 (DNMT1) was not affected by depsipeptide, a decrease in binding of DNMT1 to the promoter of these genes played a dominant role in depsipeptide-induced demethylation and reactivation. Depsipeptide also suppressed expression of histone methyltransferases G9A and SUV39H1, which in turn resulted in a decrease of di- and trimethylated H3K9 around these genes' promoter. Furthermore, both loading of heterochromatin-associated protein 1 (HP1alpha and HP1beta) to methylated H3K9 and binding of DNMT1 to these genes' promoter were significantly reduced in depsipeptide-treated cells. Similar DNA demethylation was induced by another HDAC inhibitor, apicidin, but not by trichostatin A. Our data describe a novel mechanism of HDACi-mediated DNA demethylation via suppression of histone methyltransferases and reduced recruitment of HP1 and DNMT1 to the genes' promoter.
Nuclear receptors control the function of cells by regulating transcription from specific gene networks. The establishment and maintenance of epigenetic gene marks is fundamental to the regulation of gene transcription and the control of cell function. RIP140 is a corepressor for nuclear receptors that suppresses transcription from a broad programme of metabolic genes and thereby controls energy homoeostasis in vivo. Here we show by analysis of Ucp1, a gene which is typically expressed in brown but not white adipocytes, that RIP140 is essential for both DNA and histone methylation to maintain gene repression. RIP140 expression promotes the assembly of DNA and histone methyltransferases (HMTs) on the Ucp1 enhancer and leads to methylation of specific CpG residues and histones as judged by bisulphite genomic sequencing and chromatin immunoprecipitation assays. Our results suggest that RIP140 serves as a scaffold for both DNA and HMT activities to inhibit gene transcription by two key epigenetic repression systems.
Purpose: Clinical and experimental studies have shown that diabetic retinopathy progression does not halt after termination of hyperglycemia, suggesting a "metabolic memory" phenomenon. DNA is highly dynamic, and cytosine methylation changes can last for several years. In diabetes, DNA methylation regulates expression of many genes associated with retinal mitochondrial homeostasis. Our aim was to investigate the role of DNA methylation in the metabolic memory.
Methods: Reversal of 4 days of 20 mM glucose by 4 to 8 days of 5 mM glucose, in the presence/absence of Dnmt inhibitor (5-aza-2'-deoxycytidine), was investigated on DNA methylation and its machinery in human retinal endothelial cells. The key parameters were confirmed in the retina from diabetic rats maintained in good glycemic control (glycated hemoglobin ~6%) for 3 months after 3 months of poor control (glycated hemoglobin >10%).
Results: DNA methyltransferase 1 (Dnmt 1) remained active after 4 days of normal glucose that followed 4 days of high glucose, and mtDNA stayed hypermethylated with impaired transcription. Hydroxymethylating enzyme Tet2, and matrix metalloproteinase-9 (regulated by hydroxymethylation) also remained upregulated. But, 8 days of normal glucose after 4 days of high glucose ameliorated mtDNA methylation and MMP-9 hydroxymethylation. Direct Dnmt targeting by Aza during the reversal period benefited methylation status of mtDNA and MMP-9 DNA. Similarly, reinstitution of good control after 3 months of poor control in rats did not reverse diabetes-induced increase in retinal Dnmt1 and Tet2, and alter the methylation status of mtDNA and MMP-9.
Conclusions: Retinal DNA methylation-hydroxymethylation machinery does not benefit immediately from reversal of hyperglycemia. Maintenance of good glycemic control for longer duration, and/or direct targeting DNA methylation ameliorates continuous mitochondrial damage, and could retard/halt diabetic retinopathy progression.
Background: The acute myeloid leukemia 1 (AML1)-eight-twenty-one (ETO) fusion protein generated by the t(8;21)(q22;q22) translocation is considered to display a crucial role in leukemogenesis in AML. By focusing on the anti-leukemia effects of eyes absent 4 (EYA4) gene on AML cells, we investigated the biologic and molecular mechanism associated with AML1-ETO expressed in t(8;21) AML.
Methods: Qualitative polymerase chain reaction (PCR), quantitative reverse transcription PCR (RT-PCR), and Western blotting analysis were used to observe the mRNA and protein expression levels of EYA4 in cell lines. Different plasmids (including mutant plasmids) of dual luciferase reporter vector were built to study the binding status of AML1-ETO to the promoter region of EYA4. Chromatin immunoprecipitation assay was used to study the epigenetic silencing mechanism of EYA4. Bisulfite sequencing was applied to detect the methylation status in EYA4 promoter region. The influence of EYA4 gene in the cell proliferation, apoptosis, and cell clone-forming ability was detected by the technique of Cell Counting Kit-8, flow cytometry, and clonogenic assay.
Results: EYA4 gene was hypermethylated in AML1-ETO+ patients and its expression was down-regulated by 6-fold in Kasumi-1 and SKNO-1 cells, compared to HL-60 and SKNO-1-siA/E cells, respectively. We demonstrated that AML1-ETO triggered the epigenetic silencing of EYA4 gene by binding at AML1-binding sites and recruiting histone deacetylase 1 and DNA methyltransferases. Enhanced EYA4 expression levels inhibited cellular proliferation and suppressed cell colony formation in AML1-ETO+ cell lines. We also found EYA4 transfection increased apoptosis of Kasumi-1 and SKNO-1 cells by 1.6-fold and 1.4-fold compared to negative control, respectively.
Conclusions: Our study identified EYA4 gene as targets for AML1-ETO and indicated it as a novel tumor suppressor gene. In addition, we provided evidence that EYA4 gene might be a novel therapeutic target and a potential candidate for treating AML1-ETO+ t (8;21) AML.
Purpose: Retinal mitochondria are dysfunctional in diabetes, and mitochondrial DNA (mtDNA) is damaged and its transcription is compromised. Our aim was to investigate the role of mtDNA methylation in the development of diabetic retinopathy.
Methods: Effect of high glucose (20 mM) on mtDNA methylation was analyzed in retinal endothelial cells by methylation-specific PCR and by quantifying 5-methylcytosine (5mC). Dnmt1 binding at the D-loop and Cytb regions of mtDNA was analyzed by chromatin immunoprecipitation. The role of mtDNA methylation in transcription and cell death was confirmed by quantifying transcripts of mtDNA-encoded genes (Cytb, ND6, and CoxII) and apoptosis, using cells transfected with Dnmt1-small interfering RNA (siRNA), or incubated with a Dnmt inhibitor. The key parameters were validated in the retinal microvasculature from human donors with diabetic retinopathy.
Results: High glucose increased mtDNA methylation, and methylation was significantly higher at the D-loop than at the Cytb and CoxII regions. Mitochondrial accumulation of Dnmt1 and its binding at the D-loop were also significantly increased. Inhibition of Dnmt by its siRNA or pharmacologic inhibitor ameliorated glucose-induced increase in 5mC levels and cell apoptosis. Retinal microvasculature from human donors with diabetic retinopathy presented similar increase in D-loop methylation and decrease in mtDNA transcription.
Conclusions: Hypermethylation of mtDNA in diabetes impairs its transcription, resulting in dysfunctional mitochondria and accelerated capillary cell apoptosis. Regulation of mtDNA methylation has potential to restore mitochondrial homeostasis and inhibit/retard the development of diabetic retinopathy.
Background: Melanoma is the most fatal skin cancer displaying a high degree of molecular heterogeneity. Phenotype switching is a mechanism that contributes to melanoma heterogeneity by altering transcription profiles for the transition between states of proliferation/differentiation and invasion/stemness. As phenotype switching is reversible, epigenetic mechanisms, like DNA methylation, could contribute to the changes in gene expression.
Results: Integrative analysis of methylation and gene expression datasets of five proliferative and five invasion melanoma cell cultures reveal two distinct clusters. SOX9 is methylated and lowly expressed in the highly proliferative group. SOX9 overexpression results in decreased proliferation but increased invasion in vitro. In a B16 mouse model, sox9 overexpression increases the number of lung metastases. Transcriptional analysis of SOX9-overexpressing melanoma cells reveals enrichment in epithelial to mesenchymal transition (EMT) pathways. Survival analysis of The Cancer Genome Atlas melanoma dataset shows that metastatic patients with high expression levels of SOX9 have significantly worse survival rates. Additional survival analysis on the targets of SOX9 reveals that most SOX9 downregulated genes have survival benefit for metastatic patients.
Conclusions: Our genome-wide DNA methylation and gene expression study of 10 early passage melanoma cell cultures reveals two phenotypically distinct groups. One of the genes regulated by DNA methylation between the two groups is SOX9. SOX9 induces melanoma cell invasion and metastasis and decreases patient survival. A number of genes downregulated by SOX9 have a negative impact on patient survival. In conclusion, SOX9 is an important gene involved in melanoma invasion and negatively impacts melanoma patient survival.
T-cell development in the thymus is largely controlled by an epigenetic program, involving in both DNA methylation and histone modifications. Previous studies have identified Cxxc1 as a regulator of both cytosine methylation and histone 3 lysine 4 trimethylation (H3K4me3). However, it is unknown whether Cxxc1 plays a role in thymocyte development. Here we show that T-cell development in the thymus is severely impaired in Cxxc1-deficient mice. Furthermore, we identify genome-wide Cxxc1-binding sites and H3K4me3 modification sites in wild-type and Cxxc1-deficient thymocytes. Our results demonstrate that Cxxc1 directly controls the expression of key genes important for thymocyte survival such as ROR?t and for T-cell receptor signalling including Zap70 and CD8, through maintaining the appropriate H3K4me3 on their promoters. Importantly, we show that ROR?t, a direct target of Cxxc1, can rescue the survival defects in Cxxc1-deficient thymocytes. Our data strongly support a critical role of Cxxc1 in thymocyte development.
Prostate cancer (CaP) is mostly composed of luminal-like differentiated cells, but contains a small subpopulation of basal cells (including stem-like cells), which can proliferate and differentiate into luminal-like cells. In cancers, CpG island hypermethylation has been associated with gene downregulation, but the causal relationship between the two phenomena is still debated. Here we clarify the origin and function of CpG island hypermethylation in CaP, in the context of a cancer cell hierarchy and epithelial differentiation, by analysis of separated basal and luminal cells from cancers. For a set of genes (including GSTP1) that are hypermethylated in CaP, gene downregulation is the result of cell differentiation and is not cancer specific. Hypermethylation is however seen in more differentiated cancer cells and is promoted by hyperproliferation. These genes are maintained as actively expressed and methylation-free in undifferentiated CaP cells, and their hypermethylation is not essential for either tumour development or expansion. We present evidence for the causes and the dynamics of CpG island hypermethylation in CaP, showing that, for a specific set of genes, promoter methylation is downstream of gene downregulation and is not a driver of gene repression, while gene repression is a result of tissue-specific differentiation.
We have reported previously that LIM homeobox transcription factor 1a (LMX1A) is hypermethylated and functions as a metastasis suppressor in cervical cancer cells. However, the regulation of LMX1A in carcinogenesis has not been reported. We aim to clarify whether specificity protein 1 (Sp1) and enhancer of zeste homolog 2 (EZH2) are involved in the regulation of LMX1A in cervical cancer. First we characterized the LMX1A promoter and used overexpression, knockdown, and reporter assays to show that Sp1 increased LMX1A promoter activity. Next, we used site-directed mutagenesis and electrophoresis mobility shift assays (EMSAs) to demonstrate that Sp1-binding sites were important for Sp1-mediated activation of the LMX1A promoter. Chromatin immunoprecipitation data demonstrated that Sp1 could bind directly to the LMX1A promoter and activate endogenous LMX1A expression in cells pretreated with 5-aza-2'-deoxycytidine (5-aza-dC). Knockdown of EZH2 decreased H3K27me3 histone modification but was insufficient to restore LMX1A expression. To explore the effect of EZH2 on the endogenous LMX1A promoter, we treated EZH2-knockdown cells with 5-aza-dC and trichostatin A (TSA) and then depleted the cells of drugs for 3days. H3K14ac was enriched at the LMX1A promoter in EZH2-knockdown cells and LMX1A mRNA was still expressed. Taken together, these data imply that Sp1 may activate LMX1A expression upon oncogenic stress during cervical cancer development. Moreover, suppression of EZH2 may delay resilencing of LMX1A after the removal of 5-aza-dC and TSA.
Treatment with the demethylating drugs 5-azacytidine (AZA) and decitabine (DAC) is now recognised as an effective therapy for patients with Myelodysplastic Syndromes (MDS), a range of disorders arising in clones of hematopoietic progenitor cells. A variety of cell models have been used to study the effect of these drugs on the methylation of promoter regions of tumour suppressor genes, with recent efforts focusing on the ability of these drugs to inhibit DNA methylation at low doses. However, it is still not clear how nano-molar drug treatment exerts its effects on the methylome. In this study, we have characterised changes in DNA methylation caused by prolonged low-dose treatment in a leukemic cell model (SKM-1), and present a genome-wide analysis of the effects of AZA and DAC. At nano-molar dosages, a one-month continuous treatment halved the total number of hypermethylated probes in leukemic cells and our analysis identified 803 candidate regions with significant demethylation after treatment. Demethylated regions were enriched in promoter sequences whereas gene-body CGIs were more resistant to the demethylation process. CGI methylation in promoters was strongly correlated with gene expression but this correlation was lost after treatment. Our results indicate that CGI demethylation occurs preferentially at promoters, but that it is not generally sufficient to modify expression patterns, and emphasises the roles of other means of maintaining cell state.
DNA methylation and histone deacetylation are two epigenetic mechanisms involved in the lack of estrogen receptor (ER) expression. Our previous studies demonstrated that mutant p53 along with repression complex proteins including DNMT1, HDAC1 and MeCP2 is associated with ER-negative promoter in MDA-MB-468 cells. To elucidate the molecular mechanism of estrogen receptor 1 (ESR1) gene silencing in these cells, we down-regulated DNMT1 and HDAC1 expression using siRNAs and studied the ability of DNMT1, HDAC1, MeCP2 and p53 in binding to ESR1 promoter CpG island. Our results showed that DNMT1 or HDAC1 down-regulation disassembled the repression complex proteins and mutant p53 from ER-negative promoter. The partial demethylation of ESR1 promoter and ER re-expression in down-regulated cells supports these findings. In vivo binding studies demonstrated that mutation of p53 protein in this cell line did not affect its binding capacity to DNMT1, HDAC1 and MeCP2 proteins. Our observations suggest that not only histone deacetylase activity of HDAC1 contributes to inactivation of methylated ESR1 gene but also HDAC1 presence on ESR1 promoter is important for assembly of DNMT1 in repression complex. In addition, our data revealed that mutant p53 protein binds to the promoter of ESR1 through direct interaction with HDAC1 and indirect interaction with DNMT1, MeCP2 proteins in the ER-negative MDA-MB-468 cells.
A hallmark of cancer is aberrant DNA methylation, consisting of global hypomethylation and regional hypermethylation of tumor suppressor genes. DNA methyltransferase inhibitors have been recognized as promising candidate anticancer drugs. Drug development has focused on DNA methylation inhibitors with the goal of activating tumor suppressor genes silenced by DNA methylation. 5-azacytidine (5-AC; Vidaza), a global DNA methyltransferase inhibitor, was Food and Drug Administration approved to treat myelodysplastic syndromes and is clinically tested for solid tumors. In this paper, it was demonstrated that 5'-aza-2'-deoxycytidine (5-azaCdR) activated both silenced tumor suppressor genes and pro-metastatic genes by demethylation, raising the concern that it would promote metastasis. 5-AzaCdR treatment increased the invasiveness of non-invasive breast cancer cell lines MCF-7 cells and ZR-75-1 and dramatically induced pro-metastatic genes; Urokinase plasminogen activator (uPA), matrix metalloproteinase 2 (MMP2), metastasis-associated gene (H-MTS1; S100A4) and C-X-C chemokine receptor 4 (CXCR4). The hypothesis that the blocking of cellular transformation activity of DNA methyltransferase inhibitor could be separated from the pro-metastatic activity was tested using short interfering RNA (siRNA) targeted to the different DNA methyltransferase (DNMT) genes. Although depletion of DNMT1 had the strongest effect on colony growth suppression in cellular transformation assays, it did not result in demethylation and activation of uPA, S100A4, MMP2 and CXCR4 in MCF-7 cells. Depletion of DNMT1 did not induce cellular invasion in MCF-7 and ZR-75-1 non-invasive breast cancer cell lines. These data have implications on the design of new DNA methyltransferase inhibitor and on the proper utilization of current inhibitors.
Systemic lupus erythematosus is a complex autoimmune disease caused by genetic and epigenetic alterations. DNA methylation abnormalities play an important role in systemic lupus erythematosus disease processes. MicroRNAs (miRNAs) have been implicated as fine-tuning regulators controlling diverse biological processes at the level of posttranscriptional repression. Dysregulation of miRNAs has been described in various disease states, including human lupus. Whereas previous studies have shown miRNAs can regulate DNA methylation by targeting the DNA methylation machinery, the role of miRNAs in aberrant CD4+ T cell DNA hypomethylation of lupus is unclear. In this study, by using high-throughput microRNA profiling, we identified that two miRNAs (miR-21 and miR-148a) overexpressed in CD4+ T cells from both patients with lupus and lupus-prone MRL/lpr mice, which promote cell hypomethylation by repressing DNA methyltransferase 1 (DNMT1) expression. This in turn leads to the overexpression of autoimmune-associated methylation-sensitive genes, such as CD70 and LFA-1, via promoter demethylation. Further experiments revealed that miR-21 indirectly downregulated DNMT1 expression by targeting an important autoimmune gene, RASGRP1, which mediated the Ras-MAPK pathway upstream of DNMT1; miR-148a directly downregulated DNMT1 expression by targeting the protein coding region of its transcript. Additionally, inhibition of miR-21 and miR-148a expression in CD4+ T cells from patients with lupus could increase DNMT1 expression and attenuate DNA hypomethylation. Together, our data demonstrated a critical functional link between miRNAs and the aberrant DNA hypomethylation in lupus CD4+ T cells and could help to develop new therapeutic approaches.
Epigenetic therapy of cancer using inhibitors of DNA methyltransferases (DNMT) or/and histone deacetylases (HDACs) has shown promising results in preclinical models and is being investigated in clinical trials. Homeodomain proteins play important roles in normal development and carcinogenesis. In this study, we demonstrated for the first time that an epigenetic drug could up-regulate homeobox genes in the reproductive homeobox genes on chromosome X (Rhox) family, including murine Rhox5, Rhox6, and Rhox9 and human RhoxF1 and RhoxF2 in breast, colon, and other types of cancer cells. We examined the molecular mechanisms underlining selective induction of Rhox5 in cancer cells by three epigenetic drugs: 5-aza-2'-deoxycytidine (DAC; decitabine), arsenic trioxide (ATO), and MS-275 [entinostat; N-(2-aminophenyl)-4-[N-(pyridine-3-ylmethoxy-carbonyl)aminomethyl]benzamide]. DAC induced Rhox5 mRNA expression from both distal promoter (Pd) and proximal promoter, whereas MS-275 and ATO induced gene expression from the Pd only. DAC and ATO inhibited both DNMT1 and DNMT3B protein expression, whereas MS-275 significantly reduced DNMT3B protein. In contrast to DAC, neither MS-275 nor ATO induced DNA demethylation on the Pd region. All three drugs led to enhanced acetylation of histones H3 and H4 at the promoter region. The occupancy of the activating histone mark dimethylated lysine 4 of H3 at Pd was enhanced by DAC and MS-275 but not ATO. Because they modulate gene expression with different potencies through shared and distinct epigenetic mechanisms, these epigenetic drugs may possess great potential in different applications for epigenetic therapy of cancer and other diseases.
Epigenetic inactivation of tumor suppressor genes is a common feature in human cancer. Promoter hypermethylation and histone deacetylation are reversible epigenetic mechanisms associated with transcriptional regulation. DNA methyltransferases (DNMT1 and DNMT3b) regulate and maintain promoter methylation and are overexpressed in human cancer. We performed whole-genome microarray analysis to identify genes with altered expression after RNAi-induced suppression of DNMT in a glioblastoma multiforme (GBM) cell line. We then identified genes with both decreased expression and evidence of promoter CpG island hypermethylation in GBM tissue samples using a combined whole-genome microarray transcriptome analysis in conjunction with a promoter array analysis after DNA immunoprecipitation with anti-5-methylcytidine. DNMT1 and 3b knockdown resulted in the restored expression of 308 genes that also contained promoter region hypermethylation. Of these, 43 were also found to be downregulated in GBM tissue samples. Three downregulated genes with hypermethylated promoters and restored expression in response to acute DNMT suppression were assayed for methylation changes using bisulfite sequence analysis of the promoter region after chronic DNMT suppression. Restoration of gene expression was not associated with changes in promoter region methylation, but rather with changes in histone methylation and chromatin conformation. Two of the identified genes exhibited growth suppressive activity in in vitro assays. Combining targeted genetic manipulations with comprehensive genomic and expression analyses provides a potentially powerful new approach for identifying epigenetically regulated genes in GBM.
The human gene MUC4 encodes a transmembrane mucin, ligand of ErbB2, that is associated with pancreatic tumor progression. In the normal pancreas, MUC4 is not expressed, whereas activation of its expression is observed in the early steps of pancreatic carcinogenesis. The molecular mechanisms responsible for MUC4 gene activation are however still unknown. The MUC4 5'-flanking region being GC-rich and including two CpG islands, we hypothesized that epigenetic regulation may be involved and undertook to decipher the molecular phenomenons implied. By treating cancer cell lines with 5-aza-2'-deoxycytidine (5-aza) and trichostatin A (TSA), we were able to restore MUC4 expression in a cell-specific manner. We showed by bisulfite-treated genomic DNA sequencing and chromatin immunoprecipitation that methylation of five CpG sites and establishment of a repressive histone code at the 5'-untranslated region were associated with MUC4 silencing and impaired its activation by Sp1. Direct involvement of DNMT3A, DNMT3B, HDAC1, and HDAC3 was demonstrated by RNA interference and chromatin immunoprecipitation. Moreover, inhibition of histone deacetylation by TSA was associated with strong MUC4 repression in high-expressing cells. In conclusion, this work shows for the first time the importance of epigenetics in regulating MUC4 expression and may represent a new strategy to inhibit its expression in epithelial tumors.
The establishment and maintenance of epigenetic gene silencing is fundamental to cell determination and function. The essential epigenetic systems involved in heritable repression of gene activity are the Polycomb group (PcG) proteins and the DNA methylation systems. Here we show that the corresponding silencing pathways are mechanistically linked. We find that the PcG protein EZH2 (Enhancer of Zeste homolog 2) interacts-within the context of the Polycomb repressive complexes 2 and 3 (PRC2/3)-with DNA methyltransferases (DNMTs) and associates with DNMT activity in vivo. Chromatin immunoprecipitations indicate that binding of DNMTs to several EZH2-repressed genes depends on the presence of EZH2. Furthermore, we show by bisulphite genomic sequencing that EZH2 is required for DNA methylation of EZH2-target promoters. Our results suggest that EZH2 serves as a recruitment platform for DNA methyltransferases, thus highlighting a previously unrecognized direct connection between two key epigenetic repression systems.
Purpose: DNMT1 maintains genomic DNA methylation at 5'-CpG-3' residues in somatic cells. Recent findings revealed that DNMT1 depletion causes distinct phenotypic changes in colon and gastric cancer cell lines, suggesting that the extent to which DNMT1 influences the expression of its target genes is cell-type specific. We determined the impact of DNMT1 depletion in prostate cancer derived cells on their gene expression profiles and cellular phenotype.
Materials and methods: Small interfering RNA was used to silence DNMT1 expression in prostate cancer derived PC3 cells (ATCC). The resulting cell line was validated by reverse transcriptase-polymerase chain reaction and Western blotting. Proliferation, migration and invasion assays were done in engineered cells to asses the effect of DNMT1 silencing on cellular phenotype. DNA microarrays were done to monitor changes in gene expression.
Results: Our data showed that DNMT1 loss dramatically decreased cell proliferation but significantly increased cell migratory and invasive potential. Additionally, in the limited set of genes whose expression and DNA methylation status were determined DNMT1 loss was associated with increased CDKN3 and claudin-3 expression, and also culminated in specific demethylation of Rb1 and RAR-beta promoters.
Conclusions: These results show that the genetic and phenotypic consequences of silencing DNMT1 in PC3 cells are markedly different from those in colon and gastric cancers, indicating that DNMT1 preferentially targets certain gene promoters. Our findings also suggest that decreasing DNMT1 levels or activity can potentially enhance prostate cancer cell invasiveness.
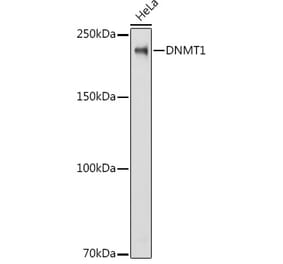
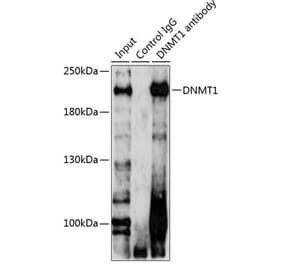
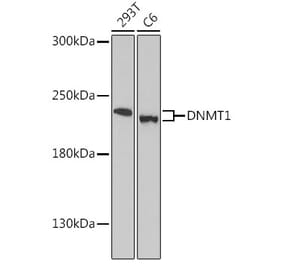
![Western Blot - Anti-Dnmt1 Antibody [ARC51348] (A307587) - Antibodies.com](https://cdn.antibodies.com/image/catalog/307/A307587_1.jpg?profile=product_alternative)
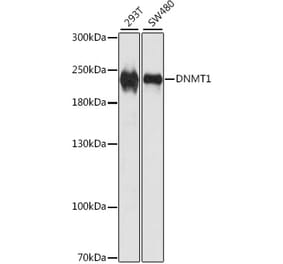
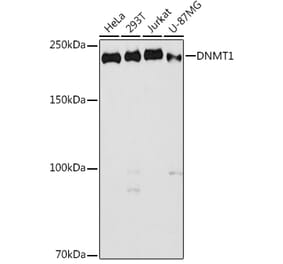
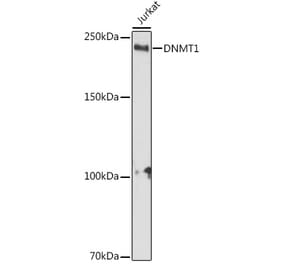
![Western Blot - Anti-DNMT1 Antibody [4G11-C7] (A304716) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304716_1.png?profile=product_alternative)
![Immunohistochemistry - Anti-DNMT1 Antibody [DNMT1/2061] (A248345) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248346_1.jpg?profile=product_alternative)
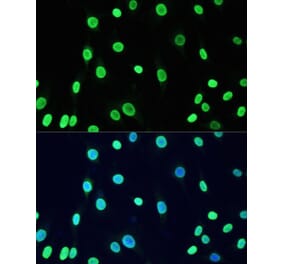
![Immunocytochemistry/Immunofluorescence - Anti-DNMT1 Antibody [11H8] (A304714) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304714_1.png?profile=product_alternative)


![Western Blot - Anti-DNMT1 Antibody [60B1220.1] (A304715) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304715_1.png?profile=product_top)
![Immunohistochemistry - Anti-DNMT1 Antibody [60B1220.1] (A304715) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304715_2.png?profile=product_top)
![Western Blot - Anti-DNMT1 Antibody [60B1220.1] (A304715) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304715_3.png?profile=product_top)
![Immunohistochemistry - Anti-DNMT1 Antibody [60B1220.1] (A304715) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304715_4.png?profile=product_top)
![Western Blot - Anti-DNMT1 Antibody [60B1220.1] (A304715) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304715_1.png?profile=product_top_thumb)
![Immunohistochemistry - Anti-DNMT1 Antibody [60B1220.1] (A304715) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304715_2.png?profile=product_top_thumb)
![Western Blot - Anti-DNMT1 Antibody [60B1220.1] (A304715) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304715_3.png?profile=product_top_thumb)
![Immunohistochemistry - Anti-DNMT1 Antibody [60B1220.1] (A304715) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304715_4.png?profile=product_top_thumb)
![Western Blot - Anti-DNMT1 Antibody [60B1220.1] (A304715) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304715_1.png?profile=product_image)
![Immunohistochemistry - Anti-DNMT1 Antibody [60B1220.1] (A304715) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304715_2.png?profile=product_image)
![Western Blot - Anti-DNMT1 Antibody [60B1220.1] (A304715) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304715_3.png?profile=product_image)
![Immunohistochemistry - Anti-DNMT1 Antibody [60B1220.1] (A304715) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304715_4.png?profile=product_image)