

Unconjugated
BACKGROUND:
Inactivation of integrin αIIbβ3 reverses platelet aggregate formation upon coagulation.
RESULTS AND CONCLUSION:
Platelets from patient (Scott) and mouse (Capn1(-/-) and Ppif(-/-)) blood reveal a dual mechanism of αIIbβ3 inactivation: by calpain-2 cleavage of integrin-associated proteins and by cyclophilin D/TMEM16F-dependent phospholipid scrambling.
SIGNIFICANCE:
These data provide novel insight into the switch mechanisms from aggregating to procoagulant platelets. Aggregation of platelets via activated integrin αIIbβ3 is a prerequisite for thrombus formation. Phosphatidylserine-exposing platelets with a key role in the coagulation process disconnect from a thrombus by integrin inactivation via an unknown mechanism. Here we show that αIIbβ3 inactivation in procoagulant platelets relies on a sustained high intracellular Ca(2+), stimulating intracellular cleavage of the β3 chain, talin, and Src kinase. Inhibition of calpain activity abolished protein cleavage, but only partly suppressed αIIbβ3 inactivation. Integrin αIIbβ3 inactivation was unchanged in platelets from Capn1(-/-) mice, suggesting a role of the calpain-2 isoform. Scott syndrome platelets, lacking the transmembrane protein TMEM16F and having low phosphatidylserine exposure, displayed reduced αIIbβ3 inactivation with the remaining activity fully dependent on calpain. In platelets from Ppif(-/-) mice, lacking mitochondrial permeability transition pore (mPTP) formation, agonist-induced phosphatidylserine exposure and αIIbβ3 inactivation were reduced. Treatment of human platelets with cyclosporin A gave a similar phenotype. Together, these data point to a dual mechanism of αIIbβ3 inactivation via calpain(-2) cleavage of integrin-associated proteins and via TMEM16F-dependent phospholipid scrambling with an assistant role of mPTP formation.



![Flow Cytometry - Anti-CD34 Antibody [QBEnd-10] - BSA and Azide free (A85654) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85659_135.jpg?profile=product_alternative)
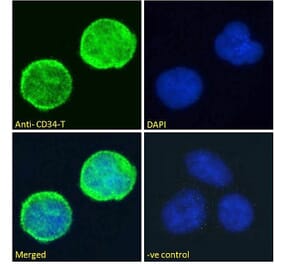
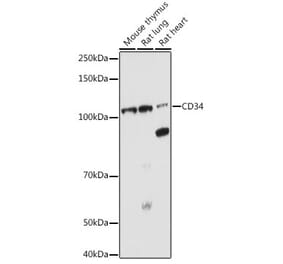
![Immunohistochemistry - Anti-CD34 Antibody [HPCA1/1171] - BSA and Azide free (A253861) - Antibodies.com](https://cdn.antibodies.com/image/catalog/253/A253861_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-CD34 Antibody [HPCA1/2598R] (A250684) - Antibodies.com](https://cdn.antibodies.com/image/catalog/250/A250684_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-CD34 Antibody [QBEnd/10] (A250677) - Antibodies.com](https://cdn.antibodies.com/image/catalog/250/A250677_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-CD34 Antibody [QBEnd/10] - BSA and Azide free (A253857) - Antibodies.com](https://cdn.antibodies.com/image/catalog/253/A253857_1.jpg?profile=product_alternative)

![Immunohistochemistry - Anti-CD34 Antibody [HPCA1/2598R] - BSA and Azide free (A253864) - Antibodies.com](https://cdn.antibodies.com/image/catalog/253/A253864_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-CD34 Antibody [HPCA1/1171] (A250681) - Antibodies.com](https://cdn.antibodies.com/image/catalog/250/A250681_1.jpg?profile=product_alternative)

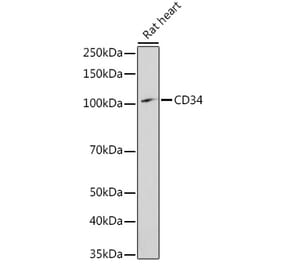
![Immunohistochemistry - Anti-CD34 Antibody [QBEnd/10 + HPCA1/763] (A250680) - Antibodies.com](https://cdn.antibodies.com/image/catalog/250/A250680_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-CD34 Antibody [QBEnd/10 + HPCA1/763] - BSA and Azide free (A253860) - Antibodies.com](https://cdn.antibodies.com/image/catalog/253/A253860_1.jpg?profile=product_alternative)