This antibody M1/70 detects CD11b (integrin alphaM subunit), a type I transmembrane protein mainly expressed on monocytes/macrophages, granulocytes and NK-cells, which associates with CD18 to form Mac-1 integrin that plays important role in cell-cell interactions.
Applications
Flow Cytometry, IP, IHC-Fr
Dilutions
Flow Cytometry: 1 µg/ml.
Reactivity
Mouse, Rabbit, Human, Non-Human Primates
Immunogen
B10 mouse spleen cells enriched for T cells.
Host
Rat
Clonality
Monoclonal
Clone ID
M1/70
Isotype
IgG2b
Conjugate
Unconjugated
Purification
Protein G chromatography.
Concentration
1 mg/ml
Product Form
Liquid
Formulation
Supplied in Phosphate Buffered Saline, pH 7.4, with 15 mM Sodium Azide.
Storage
Shipped at 4°C. Upon delivery aliquot and store at -20°C. Avoid freeze/thaw cycles.




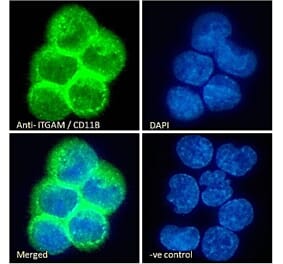

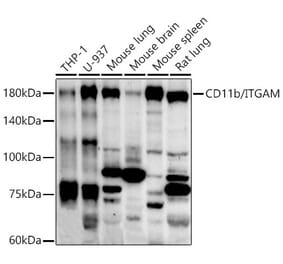
![Western Blot - Anti-CD11b Antibody [ITGAM/3338] (A249067) - Antibodies.com](https://cdn.antibodies.com/image/catalog/249/A249067_1.jpg?profile=product_alternative)
![Western Blot - Anti-CD11b Antibody [ITGAM/3338] - BSA and Azide free (A252247) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252247_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-CD11b Antibody [ITGAM/3340] (A249064) - Antibodies.com](https://cdn.antibodies.com/image/catalog/249/A249064_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-CD11b Antibody [ITGAM/3340] - BSA and Azide free (A252244) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252244_1.jpg?profile=product_alternative)
