

Anti-beta III Tubulin Antibody [TU-20] (A86691) has been discontinued and is no longer available.
View all beta III Tubulin Antibodies.
Unconjugated
Parkinson's Disease (PD) is primarily characterized by a-synuclein pathology, which manifests as intraneuronal inclusions, neuroinflammation, and neurodegeneration. However, emerging evidence also points to significant vascular impairments as a critical aspect of PD pathology, which remains largely underexplored due to the inability of traditional in vitro models to recapitulate such vascular changes. To address this unmet need, here we combine the human organ-on-a-chip technology with the principle of vasculogenic self-assembly to engineer the capillary interface of dopaminergic neurons in the substantia nigra pars compacta of the midbrain. In our proof-of-concept demonstration, we successfully recreated critical neuronal pathology in PD, including a-synuclein aggregation, inflammatory responses, and progressive neuronal degeneration, by exposing our model to specially generated PD-associated a-synuclein preformed fibrils. Importantly, this engineering approach also enables the investigation of progressive vascular changes characteristic of PD, such as endothelial dysfunction, barrier disruption, and vascular regression. Our sophisticated PD model establishes a novel platform for exploring the multifaceted nature of the disease and understanding the complex interplay between neurodegeneration and vascular pathology, offering a unique tool for developing innovative therapeutic strategies that address both the neuronal and vascular components of PD pathology.
![Immunocytochemistry - Anti-beta III Tubulin Antibody [TU-20] (A86689) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86691_816.jpg?profile=product_top)
![Immunocytochemistry - Anti-beta III Tubulin Antibody [TU-20] (A86691) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86691_817.jpg?profile=product_top)
![Immunocytochemistry - Anti-beta III Tubulin Antibody [TU-20] (A86691) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86691_818.jpg?profile=product_top)
![Immunocytochemistry - Anti-beta III Tubulin Antibody [TU-20] (A86689) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86691_816.jpg?profile=product_top_thumb)
![Immunocytochemistry - Anti-beta III Tubulin Antibody [TU-20] (A86691) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86691_817.jpg?profile=product_top_thumb)
![Immunocytochemistry - Anti-beta III Tubulin Antibody [TU-20] (A86691) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86691_818.jpg?profile=product_top_thumb)
![Immunocytochemistry - Anti-beta III Tubulin Antibody [TU-20] (A86689) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86691_816.jpg?profile=product_image)
![Immunocytochemistry - Anti-beta III Tubulin Antibody [TU-20] (A86691) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86691_817.jpg?profile=product_image)
![Immunocytochemistry - Anti-beta III Tubulin Antibody [TU-20] (A86691) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86691_818.jpg?profile=product_image)

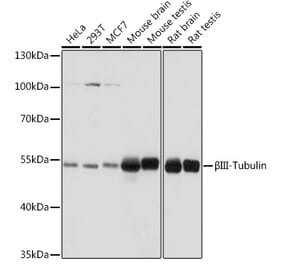
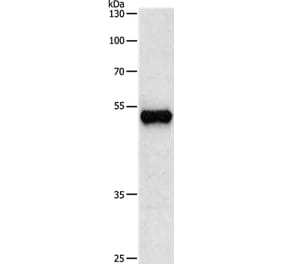
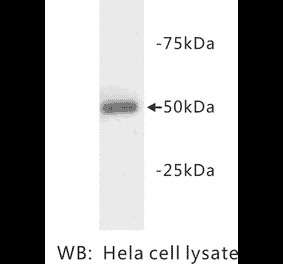
![Western Blot - Anti-beta III Tubulin Antibody [ARC0456] (A309019) - Antibodies.com](https://cdn.antibodies.com/image/catalog/309/A309019_1.jpg?profile=product_alternative)

![Immunohistochemistry - Anti-beta III Tubulin Antibody [TUBB3/3731] (A248107) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248107_1.jpg?profile=product_alternative)
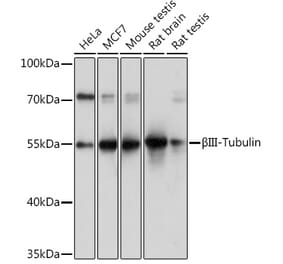

![Immunohistochemistry - Anti-beta III Tubulin Antibody [TUBB3/3731] - BSA and Azide free (A251290) - Antibodies.com](https://cdn.antibodies.com/image/catalog/251/A251290_1.jpg?profile=product_alternative)
![Protein Array - Anti-beta III Tubulin Antibody [TUBB3/3732] (A248107) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248108_1.jpg?profile=product_alternative)