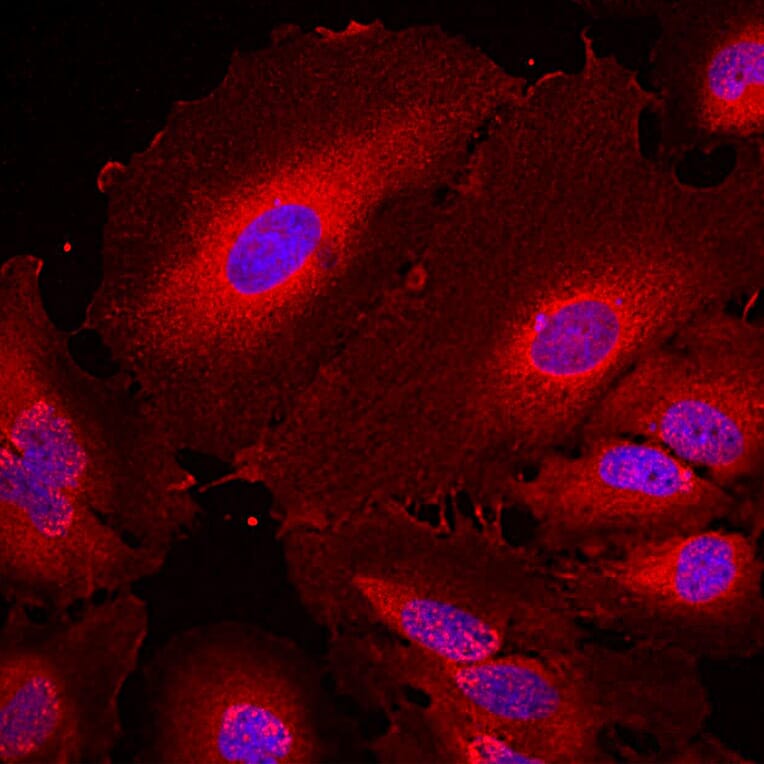
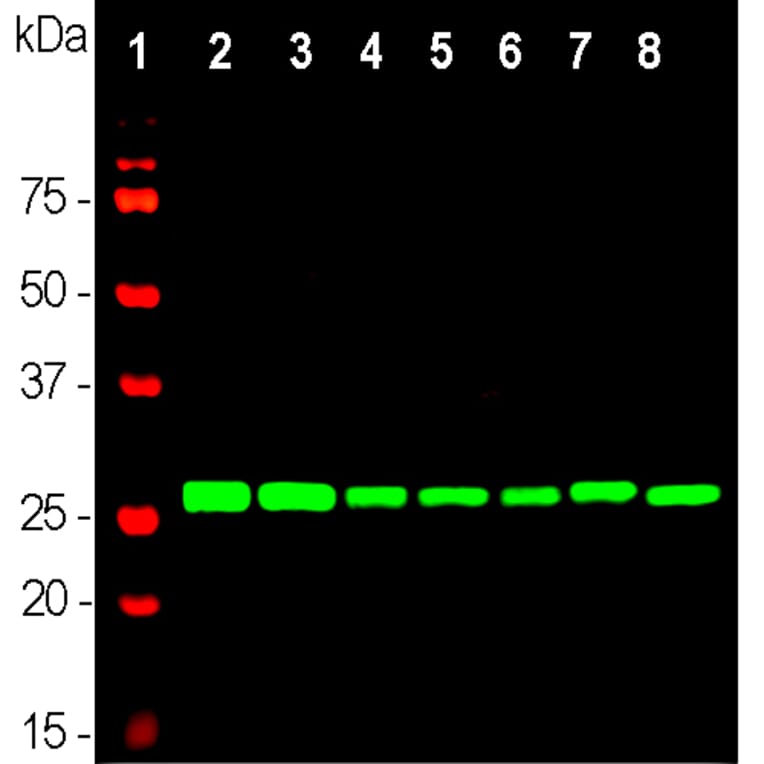
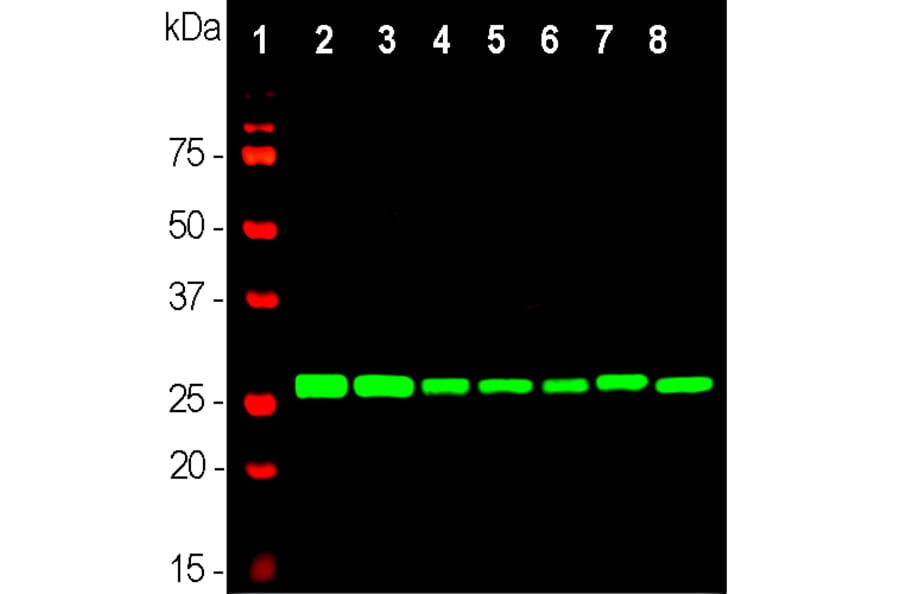
Unconjugated
High affinity nicotine-binding sites in the mammalian brain are neuronal nicotinic acetylcholine receptors (nAChR) assembled from at least alpha4 and beta2 subunits into pentameric ion channels. When exposed to ligands such as nicotine, these receptors respond by undergoing upregulation, a correlate of nicotine addiction. Upregulation can be measured using HEK293 (293) cells that stably express alpha4 and beta2 subunits using quantification of [3H]epibatidine ([3H]Eb) binding to measure mature receptors. Treatment of these cells with choline also produces upregulation through a hemicholinium3 (HC3)-sensitive (choline kinase) and an HC3-insensitive pathway which are both independent of the mechanism used by nicotine for upregulation. In both cases, upregulation is significantly enhanced by the pro-inflammatory cytokine tumor necrosis factor alpha (TNFα) which signals through its receptor Tnfr1 to activate p38Mapk. Here we report that the inhibition of class1 phosphoinositide 3-kinases isoform PI3Kbeta using the selective antagonist PI828 is alone sufficient to produce upregulation and enhance both nicotine and choline HC3-sensitive mediated upregulation. Further, these processes are impacted upon by an AG-490 sensitive Jak2-associated pathway. Both PI3Kbeta (negative) and Jak2 (positive) modulation of upregulation converge through p38Mapk and both overlap with TNFalpha enhancement of this process. Upregulation through the PI3Kbeta pathway did not require Akt. Collectively these findings support upregulation of endogenous alpha4beta2 as a balance among cellular signaling networks that are highly responsive to multiple environmental, inflammatory and metabolic agents. The findings also suggest how illness and metabolic stress could alter the expression of this important nicotinic receptor and novel avenues to intercede in modifying its expression.