+1 (314) 370-6046 or
Contact Us - Argentina
- Australia
- Austria
- Bahrain
- Belgium
- Brazil
- Bulgaria
- Cameroon
- Canada
- Chile
- China
- Colombia
- Croatia
- Cyprus
- Czech Republic
- Denmark
- Ecuador
- Egypt
- Estonia
- Finland
- France
- Germany
- Greece
- Hong Kong
- Hungary
- Iceland
- India
- Indonesia
- Iran
- Ireland
- Israel
- Italy
- Japan
- Kazakhstan
- Kuwait
- Latvia
- Lithuania
- Luxembourg
- Macedonia
- Malaysia
- Malta
- Mexico
- Monaco
- Morocco
- Netherlands
- New Zealand
- Nigeria
- Norway
- Peru
- Philippines
- Poland
- Portugal
- Qatar
- Romania
- Russia
- Saudi Arabia
- Serbia
- Singapore
- Slovakia
- Slovenia
- South Africa
- South Korea
- Spain
- Sri Lanka
- Sweden
- Switzerland
- Taiwan
- Thailand
- Turkey
- Ukraine
- UAE
- United Kingdom
- United States
- Venezuela
- Vietnam


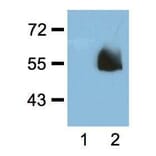
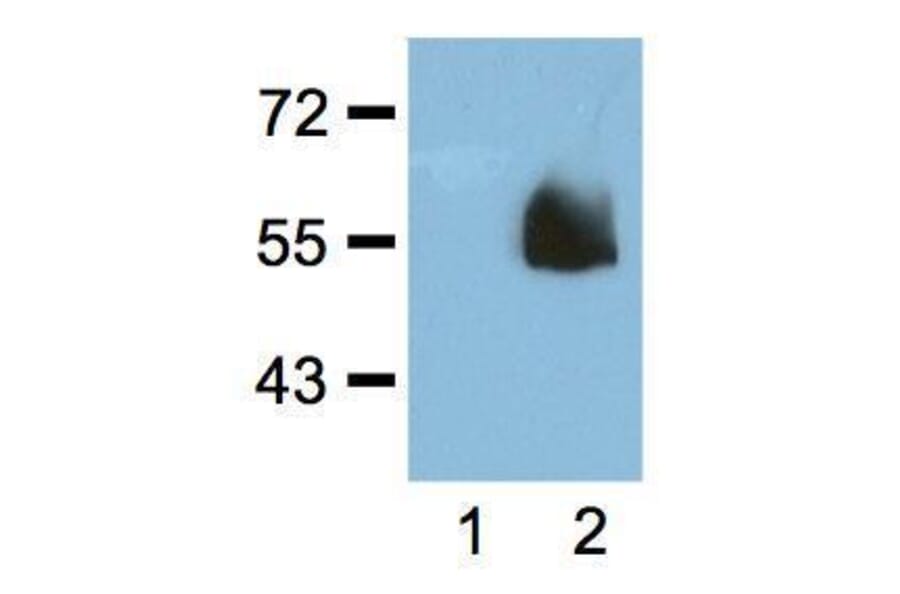

![ELISA - Anti-HA Tag Antibody [RMH02] (A121359) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121322_1.png?profile=product_alternative)
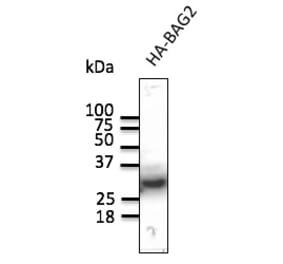

![SDS-PAGE - Anti-HA Tag Antibody [HA/279] - BSA and Azide free (A278453) - Antibodies.com](https://cdn.antibodies.com/image/catalog/278/A278453_1.jpg?profile=product_alternative)
![Western Blot - Anti-HA Tag Antibody [16.43] - BSA and Azide free (A254042) - Antibodies.com](https://cdn.antibodies.com/image/catalog/254/A254043_1.jpg?profile=product_alternative)
![Western Blot - Anti-HA Tag Antibody [16.43] (A250862) - Antibodies.com](https://cdn.antibodies.com/image/catalog/250/A250863_1.jpg?profile=product_alternative)
![SDS-PAGE - Anti-HA Tag Antibody [HA/279] (A277865) - Antibodies.com](https://cdn.antibodies.com/image/catalog/277/A277865_1.jpg?profile=product_alternative)
![Western Blot - Anti-HA Tag Antibody [RM305] (A121321) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121321_1.png?profile=product_alternative)