The epitope for this antibody is in the N-terminal region of the alpha helical coiled-coil region of GFAP, a 147 amino acid region from 71-217 of human GFAP isotype 1.
Applications
WB, ICC/IF, IHC
Dilutions
WB: 1:2,000, ICC/IF: 1:500, IHC: 1:2,000
Reactivity
Human, Rat, Mouse, Bovine, Porcine
Immunogen
GFAP isolated biochemically from pig spinal cord.
Host
Mouse
Clonality
Monoclonal
Clone ID
2A5
Isotype
IgG1
Conjugate
Unconjugated
Purification
Immunogen affinity purification.
Concentration
1 mg/ml
Molecular Weight
50 kDa
Product Form
Liquid
Formulation
Supplied in Phosphate Buffered Saline with 50% Glycerol and 5mM Sodium Azide.
Storage
Shipped at 4°C. Upon delivery aliquot and store at -20°C. Avoid freeze/thaw cycles.
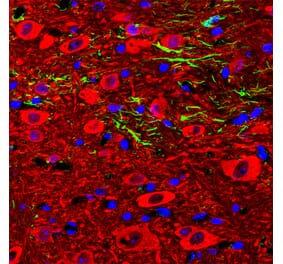
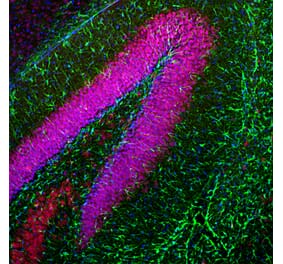
Immunofluorescent analysis of an adult rat cerebellum section stained with Anti-GFAP Antibody [2A5] (A104314), at a dilution of 1:500, in red, and co-stained with Anti-Parvalbumin Antibody (A85316), at a dilution of 1:2,000, in green. The blue is DAPI staining of nuclear DNA. Following transcardial perfusion of rat with 4% paraformaldehyde, brain was post fixed for 24 hours, and free-floating 45µM sections were stained with above antibodies. Anti-GFAP Antibody [2A5] (A104314) stains the processes of Bergmann glia and astrocytes. Anti-Parvalbumin Antibody (A85316) labels perikarya and dendrites of Purkinje cells and interneurons in the molecular layer of the cerebellum. The staining on rodent tissues is specific but not as robust as on human material.
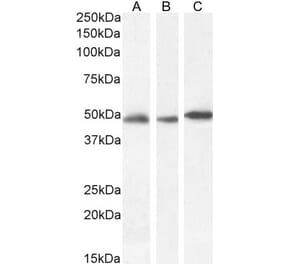
Western blot analysis of equal amount of total protein from different tissue lysates and recombinant proteins solutions using Anti-GFAP Antibody [2A5] (A104314), at a dilution of 1:2,000, in green. The lanes contain: [Lane 1] protein standard (red), [Lane 2] rat brain, [Lane 3] rat spinal cord, [Lane 4] mouse brain, [Lane 5] mouse spinal cord, [Lane 6] pig brain, [Lane 7] recombinant rat GFAP, and [Lane 8] recombinant human GFAP. Bands around 50kDa correspond to alternative transcripts and proteolytic products of GFAP. Note that Anti-GFAP Antibody [2A5] (A104314) has significantly stronger reactivity with pig and human GFAP, as compared to rodent GFAP, suggesting that it binds to an epitope which is not totally conserved across mammalian sequences.
Immunohistochemistry analysis of formalin fixed paraffin embedded histological section of human cerebellum stained with Anti-GFAP Antibody [2A5] (A104314) using the HRP/DAB staining. Counterstained with Hematoxylin (blue). Top left: region of cerebellar molecular layer containing the prominent cytoskeletal fibers of Bergmann glia which are strongly positive for GFAP. Bottom right: region of the granular layer and to further to the right is white matter, both of which contain GFAP positive astrocytes. Staining achieved with a 15 min pressure cooker heat retrieval in Antigen Retrieval Buffer, Citrate buffer at pH 6.0, and staining was performed with the Vector ImmPress rat adsorbed horse anti-mouse IgG detection kit.
Publishing research using Anti-GFAP Antibody [2A5] (A104314)? Please let us know so that we can list the citation on this page.


![Immunofluorescence - Anti-GFAP Antibody [2A5] (A104314) - Antibodies.com](https://cdn.antibodies.com/image/catalog/104/A104314_1.jpg?profile=product_top)
![Western Blot - Anti-GFAP Antibody [2A5] (A104314) - Antibodies.com](https://cdn.antibodies.com/image/catalog/104/A104314_2.jpg?profile=product_top)
![Immunohistochemistry - Anti-GFAP Antibody [2A5] (A104314) - Antibodies.com](https://cdn.antibodies.com/image/catalog/104/A104314_3.jpg?profile=product_top)
![Immunofluorescence - Anti-GFAP Antibody [2A5] (A104314) - Antibodies.com](https://cdn.antibodies.com/image/catalog/104/A104314_1.jpg?profile=product_top_thumb)
![Western Blot - Anti-GFAP Antibody [2A5] (A104314) - Antibodies.com](https://cdn.antibodies.com/image/catalog/104/A104314_2.jpg?profile=product_top_thumb)
![Immunohistochemistry - Anti-GFAP Antibody [2A5] (A104314) - Antibodies.com](https://cdn.antibodies.com/image/catalog/104/A104314_3.jpg?profile=product_top_thumb)
![Immunofluorescence - Anti-GFAP Antibody [2A5] (A104314) - Antibodies.com](https://cdn.antibodies.com/image/catalog/104/A104314_1.jpg?profile=product_image)
![Western Blot - Anti-GFAP Antibody [2A5] (A104314) - Antibodies.com](https://cdn.antibodies.com/image/catalog/104/A104314_2.jpg?profile=product_image)
![Immunohistochemistry - Anti-GFAP Antibody [2A5] (A104314) - Antibodies.com](https://cdn.antibodies.com/image/catalog/104/A104314_3.jpg?profile=product_image)
![Immunofluorescence - Anti-GFAP Antibody [5C10] (A85422) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85422_1.jpg?profile=product_alternative)
![Western Blot - Anti-GFAP Antibody [GA-5] - BSA and Azide free (A251887) - Antibodies.com](https://cdn.antibodies.com/image/catalog/251/A251887_1.jpg?profile=product_alternative)
![Western Blot - Anti-GFAP Antibody [GA-5] (A248705) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248705_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-GFAP Antibody [SPM507] - BSA and Azide free (A251888) - Antibodies.com](https://cdn.antibodies.com/image/catalog/251/A251889_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-GFAP Antibody [GA-5 + ASTRO/789] (A248708) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248709_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-GFAP Antibody [SPM248] - BSA and Azide free (A251888) - Antibodies.com](https://cdn.antibodies.com/image/catalog/251/A251888_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-GFAP Antibody [GA-5 + ASTRO/789] - BSA and Azide free (A251890) - Antibodies.com](https://cdn.antibodies.com/image/catalog/251/A251891_1.jpg?profile=product_alternative)

![Western Blot - Anti-GFAP Antibody [ARC0206] (A307282) - Antibodies.com](https://cdn.antibodies.com/image/catalog/307/A307282_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-GFAP Antibody [SPM248] (A248706) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248706_1.jpg?profile=product_alternative)