

Unconjugated
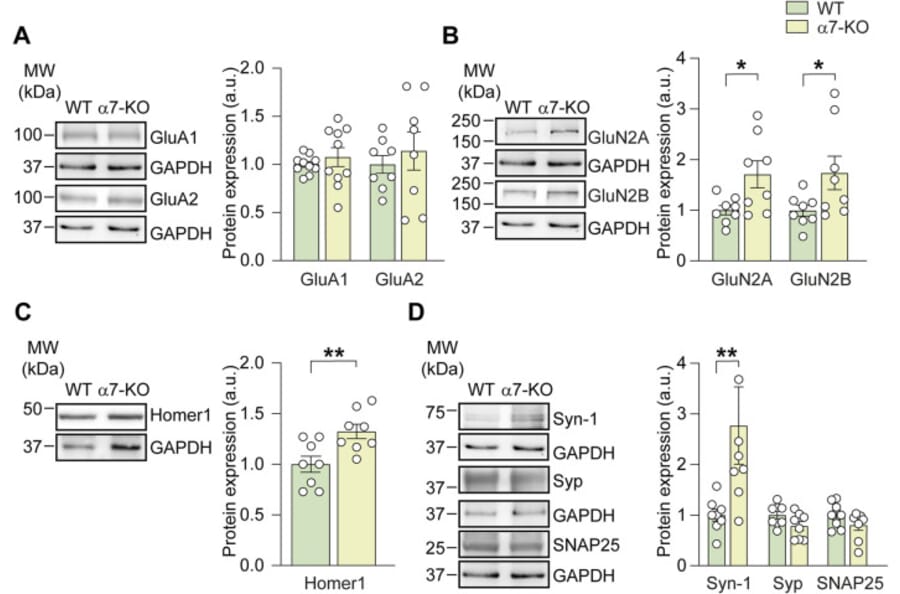
Alpha7 nicotinic acetylcholine receptors (a7-nAChRs) are ionotropic, Ca2+-permeable receptors highly expressed in brain regions involved in memory formation, such as the hippocampus. Their activation induces cation influx and neuronal depolarization, which in turn promotes glutamate release-highlighting a crucial interplay between cholinergic and glutamatergic signaling in the healthy brain. Interestingly, the genetic deletion of a7-nAChRs in mice (a7-KO mice) leads to an Alzheimer's disease (AD)-like phenotype characterized by aberrant amyloid-ß accumulation, tau phosphorylation, and neuroinflammation in aged (>12 months) mice. However, glutamatergic transmission in these mice prior to the onset of the AD-like phenotype has been poorly investigated. We thus studied molecular and functional properties of glutamatergic transmission in 4-6-months-old a7-KO mice, representing a prodromal phase of the AD-like neuropathology. We found that hippocampal CA1 neurons in brain slices from a7-KO mice showed a reduced frequency of the spontaneous excitatory post-synaptic currents (sEPSCs) compared to those of wild-type (WT) mice. On the contrary, the amplitude of sEPSCs was not affected, although in a7-KO neurons these currents displayed a longer rise time than in wild-type. CA1 neurons from a7-KO mice also exhibited a significantly smaller evoked NMDA currents than WT neurons, whereas AMPA currents were unaffected. From a molecular point of view, hippocampal neurons of a7-KO mice exhibited an increased expression of the pre-synaptic protein Synapsin-1 and of the NMDA subunits GluN2A and GluN2B, but no modifications in the expression of AMPA receptor subunits (GluA1 and GluA2) were found. Importantly, selective re-expression of the a7-nAChRs in neurons of a7-KO mice restored the evoked NMDA current amplitude and the rise time of sEPSCs, but it did not rescue the frequency of sEPSCs, thus suggesting that post-synaptic integrity depends on neuronal a7-nAChRs.
![Immunofluorescence - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_1.jpg?profile=product_top)
![Western Blot - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_2.jpg?profile=product_top)
![Immunofluorescence - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_3.jpg?profile=product_top)
![Western Blot - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_4.jpg?profile=product_top)
![Western Blot - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_5.jpg?profile=product_top)
![Western Blot - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_6.jpg?profile=product_top)
![Immunohistochemistry - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_7.jpg?profile=product_top)
![Immunofluorescence - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_1.jpg?profile=product_top_thumb)
![Western Blot - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_2.jpg?profile=product_top_thumb)
![Immunofluorescence - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_3.jpg?profile=product_top_thumb)
![Western Blot - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_4.jpg?profile=product_top_thumb)
![Western Blot - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_5.jpg?profile=product_top_thumb)
![Western Blot - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_6.jpg?profile=product_top_thumb)
![Immunohistochemistry - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_7.jpg?profile=product_top_thumb)
![Immunofluorescence - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_1.jpg?profile=product_image)
![Western Blot - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_2.jpg?profile=product_image)
![Immunofluorescence - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_3.jpg?profile=product_image)
![Western Blot - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_4.jpg?profile=product_image)
![Western Blot - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_5.jpg?profile=product_image)
![Western Blot - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_6.jpg?profile=product_image)
![Immunohistochemistry - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_7.jpg?profile=product_image)

![Western Blot - Anti-GAPDH Antibody [ARC50888] (A309068) - Antibodies.com](https://cdn.antibodies.com/image/catalog/309/A309068_1.jpg?profile=product_alternative)
![Immunocytochemistry - Anti-GAPDH Antibody [RM114] (A121303) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121396_1.png?profile=product_alternative)
