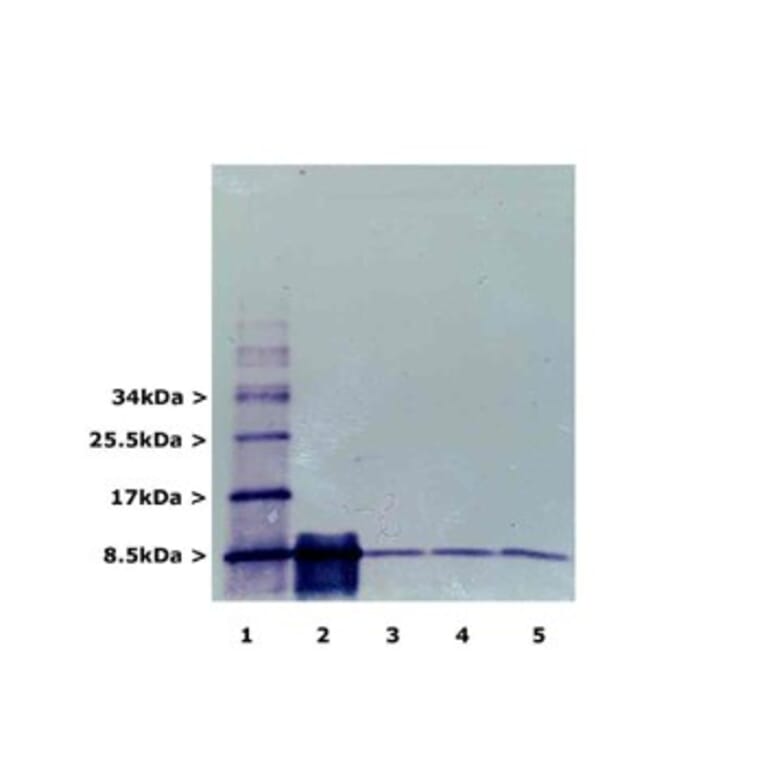
Unconjugated
Several ubiquitin chain types have remained unstudied, mainly because tools and techniques to detect these posttranslational modifications are scarce. Linkage-specific antibodies have shaped our understanding of the roles and dynamics of polyubiquitin signals but are available for only five out of eight linkage types. We here characterize K6- and K33-linkage-specific "affimer" reagents as high-affinity ubiquitin interactors. Crystal structures of affimers bound to their cognate chain types reveal mechanisms of specificity and a K11 cross-reactivity in the K33 affimer. Structure-guided improvements yield superior affinity reagents suitable for western blotting, confocal fluorescence microscopy and pull-down applications. This allowed us to identify RNF144A and RNF144B as E3 ligases that assemble K6-, K11-, and K48-linked polyubiquitin in vitro. A protocol to enrich K6-ubiquitinated proteins from cells identifies HUWE1 as a main E3 ligase for this chain type, and we show that mitofusin-2 is modified with K6-linked polyubiquitin in a HUWE1-dependent manner.
The chemical probe C60 efficiently triggers Epstein-Barr Virus (EBV) reactivation from latency through an unknown mechanism. Here, we identify the Cullin exchange factor CAND1 as a biochemical target of C60. We also identified CAND1 in an shRNA library screen for EBV lytic reactivation. Gene expression profiling revealed that C60 activates the p53 pathway and protein analysis revealed a strong stabilization and S15 phosphorylation of p53. C60 reduced Cullin1 association with CAND1 and led to a global accumulation of ubiquitylated substrates. C60 also stabilized the EBV immediate early protein ZTA through a Cullin-CAND1-interaction motif in the ZTA transcription activation domain. We propose that C60 perturbs the normal interaction and function of CAND1 with Cullins to promote the stabilization of substrates like ZTA and p53, leading to EBV reactivation from latency. Understanding the mechanism of action of C60 may provide new approaches for treatment of EBV associated tumors, as well as new tools to stabilize p53.
ESCRT proteins are implicated in myriad cellular processes, including endosome formation, fusion of autophagosomes/amphisomes with lysosomes, and apoptosis. The role played by these proteins in either facilitating or protecting against apoptosis is unclear. In this study, while trying to understand how deficiency of Mahogunin RING finger 1 (MGRN1) affects cell viability, we uncovered a novel role for its interactor, the ESCRT-I protein TSG101: it directly participates in mitigating ER stress-mediated apoptosis. The association of TSG101 with ALIX prevents predisposition to apoptosis, whereas ALIX-ALG-2 interaction favors a death phenotype. Altered Ca2+ homeostasis in cells and a simultaneous increase in the protein levels of ALIX and ALG-2 are required to elicit apoptosis by activating ER stress-associated caspase 4/12. We further demonstrate that in the presence of membrane-associated, disease-causing prion protein CtmPrP, increased ALIX and ALG-2 levels are detected along with ER stress markers and associated caspases in transgenic brain lysates and cells. These effects were rescued by overexpression of TSG101. This is significant because MGRN1 deficiency is closely associated with neurodegeneration and prenatal and neonatal mortality, which could be due to excess cell death in selected brain regions or myocardial apoptosis during embryonic development.
Within mitochondria, the ability to produce energy relies upon the architectural hallmarks of double membranes and cristae invaginations. Herein, we describe novel features of mitochondrial cristae structure, which correspond to the energetic state of the organelle. In concordance with high-energy demand, mitochondria of Drosophila indirect flight muscle exhibited extensive intra-mitochondrial membrane switches between densely packed lamellar cristae that resulted in a spiral-like cristae network and allowed for bidirectional matrix confluency. This highly interconnected architecture is expected to allow rapid equilibration of membrane potential and biomolecules across integrated regions. In addition, mutant flies with mtDNA replication defect and an accelerated aging phenotype accumulated mitochondria that contained subsections of swirling membrane alongside normal cristae. The swirling membrane had impaired energy production capacity as measured by protein composition and function. Furthermore, mitochondrial fusion and fission dynamics were affected in the prematurely aged flies. Interestingly, the normal cristae that remained in the mitochondria with swirling membranes maintained acceptable function that camouflaged them from quality control elimination. Overall, structural features of mitochondrial cristae were described in three-dimension from serial section electron tomographic analysis which reflect energetic state and mtDNA-mediated aging.
Plant architecture, a collection of genetically controlled agronomic traits, is one of the decisive factors that determine grain production. IDEAL PLANT ARCHITECTURE1 (IPA1) encodes a key transcription factor with pleiotropic effects on regulating plant architecture in rice (Oryza sativa), and IPA1 expression is controlled at the posttranscriptional level by microRNA156 and microRNA529. Here, we report the identification and characterization of IPA1 INTERACTING PROTEIN1 (IPI1), a RING-finger E3 ligase that can interact with IPA1 in the nucleus. IPI1 promotes the degradation of IPA1 in panicles, while it stabilizes IPA1 in shoot apexes. Consistent with these findings, the ipi1 loss-of-function mutants showed markedly altered plant architecture, including more tillers, enlarged panicles, and increased yield per plant. Moreover, IPI1 could ubiquitinate the IPA1-mediated complex with different polyubiquitin chains, adding K48-linked polyubiquitin chains in panicles and K63-linked polyubiquitin chains in the shoot apex. These results demonstrate that IPI1 affects plant architecture through precisely tuning IPA1 protein levels in different tissues in rice and provide new insight into the tissue-specific regulation of plant architecture and important genetic resources for molecular breeding.
Amyloid precursor protein (APP) is enriched at the synapse, but its synaptic function is still poorly understood. We previously showed that GABAergic short-term plasticity is impaired in App knock-out (App-/-) animals, but the precise mechanism by which APP regulates GABAergic synaptic transmission has remained elusive. Using electrophysiological, biochemical, moleculobiological, and pharmacological analysis, here we show that APP can physically interact with KCC2, a neuron-specific K+-Cl- cotransporter that is essential for Cl- homeostasis and fast GABAergic inhibition. APP deficiency results in significant reductions in both total and membrane KCC2 levels, leading to a depolarizing shift in the GABA reversal potential (EGABA). Simultaneous measurement of presynaptic action potentials and inhibitory postsynaptic currents (IPSCs) in hippocampal neurons reveals impaired unitary IPSC amplitudes attributable to a reduction in α1 subunit levels of GABAAR. Importantly, restoration of normal KCC2 expression and function in App-/- mice rescues EGABA, GABAAR α1 levels and GABAAR mediated phasic inhibition. We show that APP functions to limit tyrosine-phosphorylation and ubiquitination and thus subsequent degradation of KCC2, providing a mechanism by which APP influences KCC2 abundance. Together, these experiments elucidate a novel molecular pathway in which APP regulates, via protein-protein interaction with KCC2, GABAAR mediated inhibition in the hippocampus.
Oxidative stress is unavoidable for aerobic organisms. When abiotic and biotic stresses are encountered, oxidative damage could occur in cells. To avoid this damage, defense mechanisms must be timely and efficiently modulated. While the response to oxidative stress has been extensively studied in plants, little is known about how the activated response is switched off when oxidative stress is diminished. By studying Arabidopsis mutant paraquat tolerance3, we identified the genetic locus PARAQUAT TOLERANCE3 (PQT3) as a major negative regulator of oxidative stress tolerance. PQT3, encoding an E3 ubiquitin ligase, is rapidly down-regulated by oxidative stress. PQT3 has E3 ubiquitin ligase activity in ubiquitination assay. Subsequently, we identified PRMT4b as a PQT3-interacting protein. By histone methylation, PRMT4b upregulates the expression of APX1 and GPX1, encoding two key enzymes against oxidative stress. On the other hand, PRMT4b is recognized by PQT3 for targeted degradation via 26S proteasome. Therefore, we have identified PQT3 as an E3 ligase that acts as a negative regulator of activated response to oxidative stress and found that histone modification by PRMT4b at APX1 and GPX1 loci plays an important role in oxidative stress tolerance.
Methionine-1 (M1)-linked ubiquitin chains regulate the activity of NF-κB, immune homeostasis, and responses to infection. The importance of negative regulators of M1-linked chains in vivo remains poorly understood. Here, we show that the M1-specific deubiquitinase OTULIN is essential for preventing TNF-associated systemic inflammation in humans and mice. A homozygous hypomorphic mutation in human OTULIN causes a potentially fatal autoinflammatory condition termed OTULIN-related autoinflammatory syndrome (ORAS). Four independent OTULIN mouse models reveal that OTULIN deficiency in immune cells results in cell-type-specific effects, ranging from over-production of inflammatory cytokines and autoimmunity due to accumulation of M1-linked polyubiquitin and spontaneous NF-κB activation in myeloid cells to downregulation of M1-polyubiquitin signaling by degradation of LUBAC in B and T cells. Remarkably, treatment with anti-TNF neutralizing antibodies ameliorates inflammation in ORAS patients and rescues mouse phenotypes. Hence, OTULIN is critical for restraining life-threatening spontaneous inflammation and maintaining immune homeostasis.
Oncogenic mutations of the Wnt (wingless)/β-catenin pathway are frequently observed in major cancer types. Thus far, however, no therapeutic agent targeting Wnt/β-catenin signaling is available for clinical use. Here we demonstrate that axitinib, a clinically approved drug, strikingly blocks Wnt/β-catenin signaling in cancer cells, zebrafish, and Apc(min/+) mice. Notably, axitinib dramatically induces Wnt asymmetry and nonrandom DNA segregation in cancer cells by promoting nuclear β-catenin degradation independent of the GSK3β (glycogen synthase kinase3β)/APC (adenomatous polyposis coli) complex. Using a DARTS (drug affinity-responsive target stability) assay coupled to 2D-DIGE (2D difference in gel electrophoresis) and mass spectrometry, we have identified the E3 ubiquitin ligase SHPRH (SNF2, histone-linker, PHD and RING finger domain-containing helicase) as the direct target of axitinib in blocking Wnt/β-catenin signaling. Treatment with axitinib stabilizes SHPRH and thereby increases the ubiquitination and degradation of β-catenin. Our findings suggest a previously unreported mechanism of nuclear β-catenin regulation and indicate that axitinib, a clinically approved drug, would provide therapeutic benefits for cancer patients with aberrant nuclear β-catenin activation.
Pathogenic bacteria rely on secreted effector proteins to manipulate host signaling pathways, often in creative ways. CE clan proteases, specific hydrolases for ubiquitin-like modifications (SUMO and NEDD8) in eukaryotes, reportedly serve as bacterial effector proteins with deSUMOylase, deubiquitinase, or, even, acetyltransferase activities. Here, we characterize bacterial CE protease activities, revealing K63-linkage-specific deubiquitinases in human pathogens, such as Salmonella, Escherichia, and Shigella, as well as ubiquitin/ubiquitin-like cross-reactive enzymes in Chlamydia, Rickettsia, and Xanthomonas. Five crystal structures, including ubiquitin/ubiquitin-like complexes, explain substrate specificities and redefine relationships across the CE clan. Importantly, this work identifies novel family members and provides key discoveries among previously reported effectors, such as the unexpected deubiquitinase activity in Xanthomonas XopD, contributed by an unstructured ubiquitin binding region. Furthermore, accessory domains regulate properties such as subcellular localization, as exemplified by a ubiquitin-binding domain in Salmonella Typhimurium SseL. Our work both highlights and explains the functional adaptations observed among diverse CE clan proteins.
Protein ubiquitination regulated by ubiquitin ligases plays important roles in innate immunity. However, key regulators of ubiquitination during innate response and roles of new types of ubiquitination (apart from Lys48- and Lys63-linkage) in control of innate signaling have not been clearly understood. Here we report that F-box only protein Fbxo21, a functionally unknown component of SCF (Skp1-Cul1-F-box protein) complex, facilitates Lys29-linkage and activation of ASK1 (apoptosis signal-regulating kinase 1), and promotes type I interferon production upon viral infection. Fbxo21 deficiency in mice cells impairs virus-induced Lys29-linkage and activation of ASK1, attenuates c-Jun N-terminal kinase (JNK) and p38 signaling pathway, and decreases the production of proinflammatory cytokines and type I interferon, resulting in reduced antiviral innate response and enhanced virus replication. Therefore Fbxo21 is required for ASK1 activation via Lys29-linkage of ASK1 during antiviral innate response, providing mechanistic insights into non-proteolytic roles of SCF complex in innate immune response.
Mutations in leucine-rich repeat kinase 2 (LRRK2) are the most-common genetic determinants of Parkinson's disease (PD). The G2019S mutation is detected most frequently and is associated with increased kinase activity. Whereas G2019S mutant dopamine neurons exhibit neurite elongation deficits, the effect of G2019S on other neuronal subtypes is unknown. As PD patients also suffer from non-motor symptoms that may be unrelated to dopamine neuron loss, we used induced pluripotent stem cells (iPSCs) to assess morphological and functional properties of peripheral sensory neurons. LRRK2 G2019S iPSC-derived sensory neurons exhibited normal neurite length but had large microtubule-containing neurite aggregations. Additionally, LRRK2 G2019S iPSC-derived sensory neurons displayed altered calcium dynamics. Treatment with LRRK2 kinase inhibitors resulted in significant, but not complete, morphological and functional rescue. These data indicate a role for LRRK2 kinase activity in sensory neuron structure and function, which when disrupted, may lead to sensory neuron deficits in PD.
Amyotrophic lateral sclerosis is a rapidly progressing neurodegenerative disease associated with protein misfolding and aggregation. Most cases are characterized by TDP-43 positive inclusions, while a minority of familial ALS cases are instead FUS and SOD1 positive respectively. Cells can generate inclusions of variable type including previously characterized aggresomes, IPOD or JUNQ structures depending on the misfolded protein. SOD1 invariably forms JUNQ inclusions but it remains unclear whether other ALS protein aggregates arise as one of these previously described inclusion types or form unique structures. Here we show that FUS variably partitioned to IPOD, JUNQ or alternate structures, contain a mobile fraction, were not microtubule dependent and initially did not contain ubiquitin. TDP-43 inclusions formed in a microtubule independent manner, did not contain a mobile fraction but variably colocalized to JUNQ inclusions and another alternate structure. We conclude that the RNA binding proteins TDP-43 and FUS do not consistently fit the currently characterised inclusion models suggesting that cells have a larger repertoire for generating inclusions than currently thought, and imply that toxicity in ALS does not stem from a particular aggregation process or aggregate structure.
Deletion of exon 9 from Cullin-3 (CUL3, residues 403-459: CUL3(Δ403-459)) causes pseudohypoaldosteronism type IIE (PHA2E), a severe form of familial hyperkalaemia and hypertension (FHHt). CUL3 binds the RING protein RBX1 and various substrate adaptors to form Cullin-RING-ubiquitin-ligase complexes. Bound to KLHL3, CUL3-RBX1 ubiquitylates WNK kinases, promoting their ubiquitin-mediated proteasomal degradation. Since WNK kinases activate Na/Cl co-transporters to promote salt retention, CUL3 regulates blood pressure. Mutations in both KLHL3 and WNK kinases cause PHA2 by disrupting Cullin-RING-ligase formation. We report here that the PHA2E mutant, CUL3(Δ403-459), is severely compromised in its ability to ubiquitylate WNKs, possibly due to altered structural flexibility. Instead, CUL3(Δ403-459) auto-ubiquitylates and loses interaction with two important Cullin regulators: the COP9-signalosome and CAND1. A novel knock-in mouse model of CUL3(WT) (/Δ403-459) closely recapitulates the human PHA2E phenotype. These mice also show changes in the arterial pulse waveform, suggesting a vascular contribution to their hypertension not reported in previous FHHt models. These findings may explain the severity of the FHHt phenotype caused by CUL3 mutations compared to those reported in KLHL3 or WNK kinases.
The 18-kDa TSPO (translocator protein) localizes on the outer mitochondrial membrane (OMM) and participates in cholesterol transport. Here, we report that TSPO inhibits mitochondrial autophagy downstream of the PINK1-PARK2 pathway, preventing essential ubiquitination of proteins. TSPO abolishes mitochondrial relocation of SQSTM1/p62 (sequestosome 1), and consequently that of the autophagic marker LC3 (microtubule-associated protein 1 light chain 3), thus leading to an accumulation of dysfunctional mitochondria, altering the appearance of the network. Independent of cholesterol regulation, the modulation of mitophagy by TSPO is instead dependent on VDAC1 (voltage-dependent anion channel 1), to which TSPO binds, reducing mitochondrial coupling and promoting an overproduction of reactive oxygen species (ROS) that counteracts PARK2-mediated ubiquitination of proteins. These data identify TSPO as a novel element in the regulation of mitochondrial quality control by autophagy, and demonstrate the importance for cell homeostasis of its expression ratio with VDAC1.
Planar cell polarity (PCP) signaling plays a critical role in tissue morphogenesis. In mammals, disruption of three of the six "core PCP" components results in polarity-dependent defects with rotated cochlear hair cell stereocilia and open neural tube. We recently demonstrated a role of Prickle1, a core PCP molecule in Drosophila, in mammalian neuronal development. To examine Prickle1 function along a broader developmental window, we generated three mutant alleles in mice. We show that the complete loss of Prickle1 leads to systemic tissue outgrowth defects, aberrant cell organization and disruption of polarity machinery. Curiously, Prickle1 mutants recapitulate the characteristic features of human Robinow syndrome and phenocopy mouse mutants with Wnt5a or Ror2 gene defects, prompting us to explore an association of Prickle1 with the Wnt pathway. We show that Prickle1 is a proteasomal target of Wnt5a signaling and that Dvl2, a target of Wnt5a signaling, is misregulated in Prickle1 mutants. Our studies implicate Prickle1 as a key component of the Wnt-signaling pathway and suggest that Prickle1 mediates some of the WNT5A-associated genetic defects in Robinow syndrome.
The sequence of events leading to stress granule assembly in stressed cells remains elusive. We show here, using isotope labeling and ion microprobe, that proportionally more RNA than proteins are present in stress granules than in surrounding cytoplasm. We further demonstrate that the delivery of single strand polynucleotides, mRNA and ssDNA, to the cytoplasm can trigger stress granule assembly. On the other hand, increasing the cytoplasmic level of mRNA-binding proteins like YB-1 can directly prevent the aggregation of mRNA by forming isolated mRNPs, as evidenced by atomic force microscopy. Interestingly, we also discovered that enucleated cells do form stress granules, demonstrating that the translocation to the cytoplasm of nuclear prion-like RNA-binding proteins like TIA-1 is dispensable for stress granule assembly. The results lead to an alternative view on stress granule formation based on the following sequence of events: after the massive dissociation of polysomes during stress, mRNA-stabilizing proteins like YB-1 are outnumbered by the burst of nonpolysomal mRNA. mRNA freed of ribosomes thus becomes accessible to mRNA-binding aggregation-prone proteins or misfolded proteins, which induces stress granule formation. Within the frame of this model, the shuttling of nuclear mRNA-stabilizing proteins to the cytoplasm could dissociate stress granules or prevent their assembly.
New evidence indicates the involvement of protein degradation dysfunctions in neurodegeneration, innate immunity response and alcohol hepatotoxicity. We recently demonstrated that ethanol increases brain proinflammatory mediators and causes brain damage by activating Toll-like receptor 4 (TLR4) signaling in glia. However, it is uncertain if the ubiquitin-proteasome and autophagy-lysosome pathways are involved in ethanol-induced brain damage and whether the TLR4 response is implicated in proteolytic processes. Using the cerebral cortex of WT and TLR4-knockout mice with and without chronic ethanol treatment, we demonstrate that ethanol induces poly-ubiquitinated proteins accumulation and promotes immunoproteasome activation by inducing the expression of β2i, β5i and PA28α, although it decreases the 20S constitutive proteasome subunits (α2, β5). Ethanol also upregulates mTOR phosphorylation, leading to a downregulation of the autophagy-lysosome pathway (ATG12, ATG5, cathepsin B, p62, LC3) and alters the volume of autophagic vacuoles. Notably, mice lacking TLR4 receptors are protected against ethanol-induced alterations in protein degradation pathways. In summary, the present results provide the first evidence demonstrating that chronic ethanol treatment causes proteolysis dysfunctions in the mouse cerebral cortex and that these events are TLR4 dependent. These findings could provide insight into the mechanisms underlying ethanol-induced brain damage.
The activity of the ubiquitin-proteasome system, UPS, declines during aging in several multicellular organisms. The reason behind this decline remains elusive. Here, using yeast as a model system, we show that while the level and potential capacity of the 26S proteasome is maintained in replicatively aged cells, the UPS is not functioning properlyin vivo. As a consequence cytosolic UPS substrates, such as ΔssCPY* are stabilized, accumulate, and form inclusions. By integrating a pGPD-HSP104 recombinant gene into the genome, we were able to constitutively elevate protein disaggregase activity, which diminished the accumulation of protein inclusions during aging. Remarkably, this elevated disaggregation restored degradation of a 26S proteasome substrate in aged cells without elevating proteasome levels, demonstrating that age-associated aggregation obstructs UPS function. The data supports the existence of a negative feedback loop that accelerates aging by exacerbating proteostatic decline once misfolded and aggregation-prone proteins reach a critical level.
Aggregation of α-synuclein can be promoted by the tubulin polymerization-promoting protein/p25α, which we have used here as a tool to study the role of autophagy in the clearance of α-synuclein. In NGF-differentiated PC12 catecholaminergic nerve cells, we show that de novo expressed p25α co-localizes with α-synuclein and causes its aggregation and distribution into autophagosomes. However, p25α also lowered the mobility of autophagosomes and hindered the final maturation of autophagosomes by preventing their fusion with lysosomes for the final degradation of α-synuclein. Instead, p25α caused a 4-fold increase in the basal level of α-synuclein secreted into the medium. Secretion was strictly dependent on autophagy and could be up-regulated (trehalose and Rab1A) or down-regulated (3-methyladenine and ATG5 shRNA) by enhancers or inhibitors of autophagy or by modulating minus-end-directed (HDAC6 shRNA) or plus-end-directed (Rab8) trafficking of autophagosomes along microtubules. Finally, we show in the absence of tubulin polymerization-promoting protein/p25α that α-synuclein release was modulated by dominant mutants of Rab27A, known to regulate exocytosis of late endosomal (and amphisomal) elements, and that both lysosomal fusion block and secretion of α-synuclein could be replicated by knockdown of the p25α target, HDAC6, the predominant cytosolic deacetylase in neurons. Our data indicate that unconventional secretion of α-synuclein can be mediated through exophagy and that factors, which increase the pool of autophagosomes/amphisomes (e.g. lysosomal disturbance) or alter the polarity of vesicular transport of autophagosomes on microtubules, can result in an increased release of α-synuclein monomer and aggregates to the surroundings.
Osmotic homeostasis is fundamental for most cells, which face recurrent alterations of environmental osmolality that challenge cell viability. Protein damage is a consequence of hypertonic stress, but whether autophagy contributes to the osmoprotective response is unknown. Here, we investigated the possible implications of autophagy and microtubule organization on the response to hypertonic stress. We show that hypertonicity rapidly induced long-lived protein degradation, LC3-II generation and Ptdlns3K-dependent formation of LC3- and ATG12-positive puncta. Lysosomotropic agents chloroquine and bafilomycin A 1, but not nutrient deprivation or rapamycin treatment, further increased LC3-II generation, as well as ATG12-positive puncta, indicating that hypertonic stress increases autophagic flux. Autophagy induction upon hypertonic stress enhanced cell survival since cell death was increased by ATG12 siRNA-mediated knockdown and reduced by rapamycin. We additionally showed that hypertonicity induces fast reorganization of microtubule networks, which is associated with strong reorganization of microtubules at centrosomes and fragmentation of Golgi ribbons. Microtubule remodeling was associated with pericentrosomal clustering of ATG12-positive autolysosomes that colocalized with SQSTM1/p62 and ubiquitin, indicating that autophagy induced by hypertonic stress is at least partly selective. Efficient autophagy by hypertonic stress required microtubule remodeling and was DYNC/dynein-dependent as autophagosome clustering was enhanced by paclitaxel-induced microtubule stabilization and was reduced by nocodazole-induced tubulin depolymerization as well as chemical (EHNA) or genetic [DCTN2/dynactin 2 (p50) overexpression] interference of DYNC activity. The data document a general and hitherto overlooked mechanism, where autophagy and microtubule remodeling play prominent roles in the osmoprotective response.
PML, the organizer of nuclear bodies (NBs), is expressed in several isoforms designated PMLI to VII which differ in their C-terminal region due to alternative splicing of a single gene. This variability is important for the function of the different PML isoforms. PML NB formation requires the covalent linkage of SUMO to PML. Arsenic trioxide (As₂O₃) enhances PML SUMOylation leading to an increase in PML NB size and promotes its interaction with RNF4, a poly-SUMO-dependent ubiquitin E3 ligase responsible for proteasome-mediated PML degradation. Furthermore, the presence of a bona fide SUMO Interacting Motif (SIM) within the C-terminal region of PML seems to be required for recruitment of other SUMOylated proteins within PML NBs. This motif is present in all PML isoforms, except in the nuclear PMLVI and in the cytoplasmic PMLVII. Using a bioluminescence resonance energy transfer (BRET) assay in living cells, we found that As₂O₃ enhanced the SUMOylation and interaction with RNF4 of nuclear PML isoforms (I to VI). In addition, among the nuclear PML isoforms, only the one lacking the SIM sequence, PMLVI, was resistant to As₂O₃-induced PML degradation. Similarly, mutation of the SIM in PMLIII abrogated its sensitivity to As₂O₃-induced degradation. PMLVI and PMLIII-SIM mutant still interacted with RNF4. However, their resistance to the degradation process was due to their inability to be polyubiquitinated and to recruit efficiently the 20S core and the β regulatory subunit of the 11S complex of the proteasome in PML NBs. Such resistance of PMLVI to As₂O₃-induced degradation was alleviated by overexpression of RNF4. Our results demonstrate that the SIM of PML is dispensable for PML SUMOylation and interaction with RNF4 but is required for efficient PML ubiquitination, recruitment of proteasome components within NBs and proteasome-dependent degradation of PML in response to As₂O₃.
Macrophage activation, including classical (M1) activation and alternative (M2) activation, plays important roles in host immune response and pathogenesis of diseases. Ubiquitination has been shown to be involved in the differentiation of immune cells and in the regulation of immune responses. However, the role of ubiquitination during M1 versus M2 polarization is poorly explored. Here, we showed that arginase 1 (Arg1), a well recognized marker of M2 macrophages, is highly up-regulated in peritoneal macrophages derived from E3 ubiquitin ligase Nrdp1 transgenic (Nrdp1-TG) mice. Furthermore, other M2 feature markers such as MR, Ym1, and Fizz1, as well as Th2 cytokine IL-10, are also up-regulated in Nrdp1-TG macrophages after IL-4 stimulation. Knockdown of Nrdp1 expression effectively inhibits IL-4-induced expression of M2-related genes in macrophages. Moreover, Nrdp1 inhibits LPS-induced production of inducible NOS and pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 in macrophages. Immunoprecipitation assays show that Nrdp1 interacts with and ubiquitinates transcriptional factor C/EBPβ via Lys-63-linked ubiquitination. Nrdp1 enhances C/EBPβ-triggered transcriptional activation of the Arg1 reporter gene in the presence of IL-4 stimulation. Thus, we demonstrate that Nrdp1-mediated ubiquitination and activation of C/EBPβ contributes to a ubiquitin-dependent nonproteolytic pathway that up-regulates Arg1 expression and promotes M2 macrophage polarization.
(Macro)Autophagy is a phylogenetically conserved membrane-trafficking process that functions to deliver cytoplasmic cargoes to lysosomes for digestion. The process is a major mechanism for turnover of cellular constituents and is therefore critical for maintaining cellular homeostasis. Macroautophagy is characteristically distinct from other forms of autophagy due to the formation of double-membraned vesicles termed autophagosomes which encapsulate cargoes prior to fusion with lysosomes. Autophagosomes contain an integral membrane-bound form (LC3-II) of the microtubule-associated protein 1 light chain 3 β (MAP1LC3B), which has become a gold-standard marker to detect accumulation of autophagosomes and thereby changes in macroautophagy. Due to the role played by macroautophagy in various diseases, the detection of autophagosomes in tissue sections is frequently desired. To date, however, the detection of endogenous LC3-II on paraffin-embedded tissue sections has proved problematic. We report here a simple, optimized and validated method for the detection of LC3-II by immunohistochemistry in human and mouse tissue samples that we believe will be a useful resource for those wishing to study macroautophagy ex vivo.
Sanfilippo syndrome type B (MPS IIIB) is characterized by profound mental retardation in childhood, dementia and death in late adolescence; it is caused by deficiency of α-N-acetylglucosaminidase and resulting lysosomal storage of heparan sulfate. A mouse model, generated by homologous recombination of the Naglu gene, was used to study pathological changes in the brain. We found earlier that neurons in the medial entorhinal cortex (MEC) and the dentate gyrus showed a number of secondary defects, including the presence of hyperphosphorylated tau (Ptau) detected with antibodies raised against Ptau in Alzheimer disease brain. By further use of immunohistochemistry, we now show staining in neurons of the same area for beta amyloid, extending the resemblance to Alzheimer disease. Ptau inclusions in the dentate gyrus of MPS IIIB mice were reduced in number when the mice were administered LiCl, a specific inhibitor of Gsk3β. Additional proteins found elevated in MEC include proteins involved in autophagy and the heparan sulfate proteoglycans, glypicans 1 and 5, the latter closely related to the primary defect. The level of secondary accumulations was associated with elevation of glypican, as seen by comparing brains of mice at different ages or with different mucopolysaccharide storage diseases. The MEC of an MPS IIIA mouse had the same intense immunostaining for glypican 1 and other markers as MPS IIIB, while MEC of MPS I and MPS II mice had weak staining, and MEC of an MPS VI mouse had no staining at all for the same proteins. A considerable amount of glypican was found in MEC of MPS IIIB mice outside of lysosomes. We propose that it is the extralysosomal glypican that would be harmful to neurons, because its heparan sulfate branches could potentiate the formation of Ptau and beta amyloid aggregates, which would be toxic as well as difficult to degrade.
Stil (Sil, SCL/TAL1 interrupting locus) is a cytosolic and centrosomal protein expressed in proliferating cells that is required for mouse and zebrafish neural development and is mutated in familial microcephaly. Recently the Drosophila melanogaster ortholog of Stil was found to be important for centriole duplication. Consistent with this finding, we report here that mouse embryonic fibroblasts lacking Stil are characterized by slow growth, low mitotic index and absence of clear centrosomes. We hypothesized that Stil regulates mitosis through the tumor suppressor Chfr, an E3 ligase that blocks mitotic entry in response to mitotic stress. Mouse fibroblasts lacking Stil by genomic or RNA interference approaches, as well as E9.5 Stil(-/-) embryos, express high levels of the Chfr protein and reduced levels of the Chfr substrate Plk1. Exogenous expression of Stil, knockdown of Chfr or overexpression of Plk1 reverse the abnormal mitotic phenotypes of fibroblasts lacking Stil. We further demonstrate that Stil increases Chfr auto-ubiquitination and reduces its protein stability. Thus, Stil is required for centrosome organization, entry into mitosis and cell proliferation, and these functions are at least partially mediated by Chfr and its targets. This is the first identification of a negative regulator of the Chfr mitotic checkpoint.
Ubiquitination and proteasome-mediated degradation of proteins are crucial for eukaryotic physiology and development. The largest class of E3 ubiquitin ligases is made up of the cullin-RING ligases (CRLs), which themselves are positively regulated through conjugation of the ubiquitin-like peptide RUB/NEDD8 to cullins. RUB modification is antagonized by the COP9 signalosome (CSN), an evolutionarily conserved eight-subunit complex that is essential in most eukaryotes and cleaves RUB from cullins. The CSN behaves genetically as an activator of CRLs, although it abolishes CRL activity in vitro. This apparent paradox was recently reconciled in different organisms, as the CSN was shown to prevent autocatalytic degradation of several CRL substrate adaptors. We tested for such a mechanism in the model plant Arabidopsis by measuring the impact of a newly identified viable csn2 mutant on the activity and stability of SCF(TIR1), a receptor to the phytohormone auxin and probably the best characterized plant CRL. Our analysis reveals that not only the F-box protein TIR1 but also relevant cullins are destabilized in csn2 and other Arabidopsis csn mutants. These results provide an explanation for the auxin resistance of csn mutants. We further observed in vivo a post-translational modification of TIR1 dependent on the proteasome inhibitor MG-132 and provide evidence for proteasome-mediated degradation of TIR1, CUL1, and ASK1 (Arabidopsis SKP1 homolog). These results are consistent with CSN-dependent protection of Arabidopsis CRLs from autocatalytic degradation, as observed in other eukaryotes, and provide evidence for antagonist roles of the CSN and 26S proteasome in modulating accumulation of the plant CRL SCF(TIR1).
Both p53 and its repressor Mdm2 are subject to ubiquitination and proteasomal degradation. We show that knockdown of the deubiquitinating enzyme USP5 (isopeptidase T) results in an increase in the level and transcriptional activity of p53. Suppression of USP5 stabilizes p53, whereas it has little or no effect on the stability of Mdm2. This provides a mechanism for transcriptional activation of p53. USP5 knockdown interferes with the degradation of ubiquitinated p53 rather than attenuating p53 ubiquitination. In vitro studies have shown that a preferred substrate for USP5 is unanchored polyubiquitin. Consistent with this, we observed for the first time in a mammalian system that USP5 makes a major contribution to Lys-48-linked polyubiquitin disassembly and that suppression of USP5 results in the accumulation of unanchored polyubiquitin chains. Ectopic expression of a C-terminal mutant of ubiquitin (G75A/G76A), which also causes the accumulation of free polyubiquitin, recapitulates the effects of USP5 knockdown on the p53 pathway. We propose a model in which p53 is selectively stabilized because the unanchored polyubiquitin that accumulates after USP5 knockdown is able to compete with ubiquitinated p53 but not with Mdm2 for proteasomal recognition. This raises the possibility that there are significant differences in proteasomal recognition of p53 and Mdm2. These differences could be exploited therapeutically. Our study reveals a novel mechanism for regulation of p53 and identifies USP5 as a potential target for p53 activating therapeutic agents for the treatment of cancer.
Nod1 and Nod2 are intracellular proteins that are involved in host recognition of specific bacterial molecules and are genetically associated with several inflammatory diseases. Nod1 and Nod2 stimulation activates NF-kappaB through RICK, a caspase-recruitment domain-containing kinase. However, the mechanism by which RICK activates NF-kappaB in response to Nod1 and Nod2 stimulation is unknown. Here we show that RICK is conjugated with lysine-63-linked polyubiquitin chains at lysine 209 (K209) located in its kinase domain upon Nod1 or Nod2 stimulation and by induced oligomerization of RICK. Polyubiquitination of RICK at K209 was essential for RICK-mediated IKK activation and cytokine/chemokine secretion. However, RICK polyubiquitination did not require the kinase activity of RICK or alter the interaction of RICK with NEMO, a regulatory subunit of IkappaB kinase (IKK). Instead, polyubiquitination of RICK was found to mediate the recruitment of TAK1, a kinase that was found to be essential for Nod1-induced signaling. Thus, RICK polyubiquitination links TAK1 to IKK complexes, a critical step in Nod1/Nod2-mediated NF-kappaB activation.
The family of cytoplasmic Janus (Jak) tyrosine kinases plays an essential role in cytokine signal transduction, regulating cell survival and gene expression. Ligand-induced receptor dimerization results in phosphorylation of Jak2 on activation loop tyrosine Y1007 and stimulation of its catalytic activity, which, in turn, results in activation of several downstream signaling cascades. Recently, the catalytic activity of Jak2 has been found to be subject to negative regulation through various mechanisms including association with SOCS proteins. Here we show that the ubiquitin-dependent proteolysis pathway is involved in the regulation of the turnover of activated Jak2. In unstimulated cells Jak2 was monoubiquitinated, and interleukin-3 or gamma interferon stimulation induced polyubiquitination of Jak2. The polyubiquitinated Jak2 was rapidly degraded through proteasomes. By using different Jak2 mutants we show that tyrosine-phosphorylated Jak2 is preferentially polyubiquitinated and degraded. Furthermore, phosphorylation of Y1007 on Jak2 was required for proteasomal degradation and for SOCS-1-mediated downregulation of Jak2. The proteasome inhibitor treatment stabilized the Jak2-SOCS-1 protein complex and inhibited the proteolysis of Jak2. In summary, these results indicate that the ubiquitin-proteasome pathway negatively regulates tyrosine-phosphorylated Jak2 in cytokine receptor signaling, which provides an additional mechanism to control activation of Jak2 and maintain cellular homeostasis.
BACKGROUND:
Recent reports demonstrate that multiple forms of cardiovascular stress, including pressure overload, chronic ischemia, and infarction-reperfusion injury, provoke an increase in autophagic activity in cardiomyocytes. However, nothing is known regarding molecular events that stimulate autophagic activity in stressed myocardium. Because autophagy is a highly conserved process through which damaged proteins and organelles can be degraded, we hypothesized that stress-induced protein aggregation is a proximal trigger of cardiomyocyte autophagy.
METHODS AND RESULTS:
Here, we report that pressure overload promotes accumulation of ubiquitinated protein aggregates in the left ventricle, development of aggresome-like structures, and a corresponding induction of autophagy. To test for causal links, we induced protein accumulation in cultured cardiomyocytes by inhibiting proteasome activity, finding that aggregation of polyubiquitinated proteins was sufficient to induce cardiomyocyte autophagy. Furthermore, attenuation of autophagic activity dramatically enhanced both aggresome size and abundance, consistent with a role for autophagic activity in protein aggregate clearance.
CONCLUSIONS:
We conclude that protein aggregation is a proximal trigger of cardiomyocyte autophagy and that autophagic activity functions to attenuate aggregate/aggresome formation in heart. Findings reported here are the first to demonstrate that protein aggregation occurs in response to hemodynamic stress, situating pressure-overload heart disease in the category of proteinopathies.
In this study, we investigated the development of endoplasmic reticulum (ER) stress after traumatic brain injury (TBI) and the efficacy of post-TBI administration of docosahexaenoic acid (DHA) in reducing ER stress. TBI was induced by cortical contusion injury in Sprague-Dawley rats. Either DHA (16 mg/kg in DMSO) or vehicle DMSO (1 ml/kg) was administered intraperitoneally at 5 min after TBI, followed by a daily dose for 3-21 d. TBI triggered sustained expression of the ER stress marker proteins including phosphorylated eukaryotic initiation factor-2α, activating transcription factor 4, inositol requiring kinase 1, and C/EBP homologous protein in the ipsilateral cortex at 3-21 d after TBI. The prolonged ER stress was accompanied with an accumulation of abnormal ubiquitin aggregates and increased expression of amyloid precursor protein (APP) and phosphorylated tau (p-Tau) in the frontal cortex after TBI. The ER stress marker proteins were colocalized with APP accumulation in the soma. Interestingly, administration of DHA attenuated all ER stress marker proteins and reduced the accumulation of both ubiquitinated proteins and APP/p-Tau proteins. In addition, the DHA-treated animals exhibited early recovery of their sensorimotor function after TBI. In summary, our study demonstrated that TBI induces a prolonged ER stress, which is positively correlated with abnormal APP accumulation. The sustained ER stress may play a role in chronic neuronal damage after TBI. Our findings illustrate that post-TBI administration of DHA has therapeutic potentials in reducing ER stress, abnormal protein accumulation, and neurological deficits.
C-Jun is a major transcription factor belonging to the activating protein 1 (AP-1) family. Phosphorylation has been shown to be critical for c-Jun activation and stability. Here, we report that Jra, the Drosophila Jun protein, is acetylated in vivo. We demonstrate that the acetylation of Jra leads to its rapid degradation in response to osmotic stress. Intriguingly, we also found that Jra phosphorylation antagonized its acetylation, indicating the opposite roles of acetylation and phosphorylation in Jra degradation process under osmotic stress. Our results provide new insights into how c-Jun proteins are precisely regulated by the interplay of different posttranslational modifications.
DNA methylation controls many aspects of plant growth and development. Here, we report a novel annual growth potential change that may correlate with changes in levels of the major DNA demethylases and methyltransferases in cotton ovules harvested at different times of the year. The abundances of DNA demethylases, at both the mRNA and protein levels, increased significantly from February to August and decreased during the remainder of the 12-month period, with the opposite pattern observed for DNA methyltransferases. Over the course of one year, substantial changes in methylcytosine content was observed at certain CHH sites (H = A, C, or T) in the promoter regions of the ETHYLENE RESPONSIVE FACTOR 6 (ERF6), SUPPRESSION OF RVS 161 DELTA 4 (SUR4) and 3-KETOACYL-COA SYNTHASE 13 (KCS13), which regulate cotton fiber growth. Three independent techniques were used to confirm the annual fluctuations in DNA methylation. Furthermore, in homozygous RNAi lines specifically targeting REPRESSOR OF SILENCING 1 (ROS1, a conserved DNA demethylase domain), promotion of DNA methylation significantly reduced fiber growth during August.
The E3 ubiquitin ligase Mule/ARF-BP1 plays an important role in the cellular DNA damage response by controlling base excision repair and p53 protein levels. However, how the activity of Mule is regulated in response to DNA damage is currently unknown. Here, we report that the Ser18-containing isoform of the USP7 deubiquitylation enzyme (USP7S) controls Mule stability by preventing its self-ubiquitylation and subsequent proteasomal degradation. We find that in response to DNA damage, downregulation of USP7S leads to self-ubiquitylation and proteasomal degradation of Mule, which eventually leads to p53 accumulation. Cells that are unable to downregulate Mule show reduced ability to upregulate p53 levels in response to DNA damage. We also find that, as Mule inactivation is required for stabilization of base excision repair enzymes, the failure of cells to downregulate Mule after DNA damage results in deficient DNA repair. Our data describe a novel mechanism by which Mule is regulated in response to DNA damage and coordinates cellular DNA damage responses and DNA repair.
A missense mutation in the alphaB-crystallin (CryAB) gene triggers a severe form of desmin-related cardiomyopathy (DRCM) characterized by accumulation of misfolded proteins. We hypothesized that autophagy increases in response to protein aggregates and that this autophagic activity is adaptive. Mutant CryAB (CryAB(R120G)) triggered a >2-fold increase in cardiomyocyte autophagic activity, and blunting autophagy increased the rate of aggregate accumulation and the abundance of insoluble CryAB(R120G)-associated aggregates. Cardiomyocyte-restricted overexpression of CryAB(R120G) in mice induced intracellular aggregate accumulation and systolic heart failure by 12 months. As early as 2 months (well before the earliest declines in cardiac function), we detected robust autophagic activity. To test the functional significance of autophagic activation, we crossed CryAB(R120G) mice with animals harboring heterozygous inactivation of beclin 1, a gene required for autophagy. Blunting autophagy in vivo dramatically hastened heart failure progression with a 3-fold increase in interstitial fibrosis, greater accumulation of polyubiquitinated proteins, larger and more extensive intracellular aggregates, accelerated ventricular dysfunction, and early mortality. This study reports activation of autophagy in DRCM. Further, our findings point to autophagy as an adaptive response in this proteotoxic form of heart disease.
Amyotrophic lateral sclerosis (ALS) is a motor neuron disease that leads to loss of motor function and early death. About 5% of cases are inherited, with the majority of identified linkages in the gene encoding copper, zinc-superoxide dismutase (SOD1). Strong evidence indicates that the SOD1 mutations confer dominant toxicity on the protein. To provide new insight into mechanisms of ALS, we have generated and characterized a model for familial ALS in Drosophila with transgenic expression of human SOD1. Expression of wild type or disease-linked (A4V, G85R) mutants of human SOD1 selectively in motor neurons induced progressive climbing deficits. These effects were accompanied by defective neural circuit electrophysiology, focal accumulation of human SOD1 protein in motor neurons, and a stress response in surrounding glia. However, toxicity was not associated with oligomerization of SOD1 and did not lead to neuronal loss. These studies uncover cell-autonomous injury by SOD1 to motor neurons in vivo, as well as non-autonomous effects on glia, and provide the foundation for new insight into injury and protection of motor neurons in ALS.
The proinflammatory cytokine tumor necrosis factor (TNF) alpha signals both cell survival and death. The biological outcome of TNFalpha treatment is determined by the balance between NF-kappaB and Jun kinase (JNK) signaling; NF-kappaB promotes survival, whereas JNK enhances cell death. Critically, identity of a JNK substrate that promotes TNFalpha-induced apoptosis has been outstanding. Here we show that TNFalpha-mediated JNK activation accelerates turnover of the NF-kappaB-induced antiapoptotic protein c-FLIP, an inhibitor of caspase-8. This is not due to direct c-FLIP phosphorylation but depends on JNK-mediated phosphorylation and activation of the E3 ubiquitin ligase Itch, which specifically ubiquitinates c-FLIP and induces its proteasomal degradation. JNK1 or Itch deficiency or treatment with a JNK inhibitor renders mice resistant in three distinct models of TNFalpha-induced acute liver failure, and cells from these mice do not display inducible c-FLIP(L) ubiquitination and degradation. Thus, JNK antagonizes NF-kappaB during TNFalpha signaling by promoting the proteasomal elimination of c-FLIP(L).
One of the key pathological hallmarks of Alzheimer disease (AD) is the accumulation of paired helical filaments (PHFs) of hyperphosphorylated microtubule-associated protein Tau. Tandem mass spectrometry was employed to examine PHF-Tau post-translational modifications, in particular protein phosphorylation and ubiquitination, to shed light on their role in the early stages of Alzheimer disease. PHF-Tau from Alzheimer disease brain was affinity-purified by MC1 monoclonal antibody to isolate a soluble fraction of PHF-Tau in a conformation unique to human AD brain. A large number of phosphorylation sites were identified by employing a data-dependent neutral loss algorithm to trigger MS3 scans of phosphopeptides. It was found that soluble PHF-Tau is ubiquitinated at its microtubule-binding domain at residues Lys-254, Lys-311, and Lys-353, suggesting that ubiquitination of PHF-Tau may be an earlier pathological event than previously thought and that ubiquitination could play a regulatory role in modulating the integrity of microtubules during the course of AD. Tandem mass spectrometry data for ubiquitin itself indicate that PHF-Tau is modified by three polyubiquitin linkages, at Lys-6, Lys-11, and Lys-48. Relative quantitative analysis indicates that Lys-48-linked polyubiquitination is the primary form of polyubiquitination with a minor portion of ubiquitin linked at Lys-6 and Lys-11. Because modification by Lys-48-linked polyubiquitin chains is known to serve as the essential means of targeting proteins for degradation by the ubiquitin-proteasome system, and it has been reported that modification at Lys-6 inhibits ubiquitin-dependent protein degradation, a failure of the ubiquitin-proteasome system could play a role in initiating the formation of degradation-resistant PHF tangles.
During neuronal maturation, dendrites develop from immature neurites into mature arbors. In response to changes in the environment, dendrites from certain mature neurons can undergo large-scale morphologic remodeling. Here, we show a group of Drosophila peripheral sensory neurons, the class IV dendritic arborization (C4da) neurons, that completely degrade and regrow their elaborate dendrites. Larval dendrites of C4da neurons are first severed from the soma and subsequently degraded during metamorphosis. This process is controlled by both intracellular and extracellular mechanisms: The ecdysone pathway and ubiquitin-proteasome system (UPS) are cell-intrinsic signals that initiate dendrite breakage, and extracellular matrix metalloproteases are required to degrade the severed dendrites. Surprisingly, C4da neurons retain their axonal projections during concurrent dendrite degradation, despite activated ecdysone and UPS pathways. These results demonstrate that, in response to environmental changes, certain neurons have cell-intrinsic abilities to completely lose their dendrites but keep their axons and subsequently regrow their dendritic arbors.
Here we demonstrate a novel p53-independent interaction between the nucleolar tumor suppressors, p14 Arf and Werners helicase (WRN). Binding of p14 Arf to WRN is multivalent and resembles the binding of p14 Arf to Mdm2. Residues 2-14 and 82-101 of p14 Arf and residues in the central region and C terminus of WRN have particular importance for binding. p14 Arf promotes small ubiquitin-like modifier (SUMO) modification of WRN in a synergistic manner with the SUMO-conjugating enzyme, UBCH9. p14 Arf causes redistribution of WRN within the nucleus, and this effect is reversed by expression of a SUMO-specific protease, thus implicating the SUMO conjugation pathway in WRN re-localization. We establish that the ability to promote SUMO conjugation is a general property of the p14 Arf tumor suppressor.
To study the role of Abeta amyloid deposits in the generation of cytoskeletal lesions, we have generated a transgenic mouse line coexpressing in the same neurons a wild-type human tau isoform (0N3R), a mutant form of APP (751SL) and a mutant form of PS1 (M146L). These mice developed early cerebral extracellular deposits of Abeta, starting at 2.5 months. A somatodendritic neuronal accumulation of transgenic tau protein was observed in tau only and in tau/PS1/APP transgenic mice, including in neurons adjacent to Abeta deposits. The phosphorylation status of this somatodendritic tau was similar in the two transgenic lines. The Abeta deposits were surrounded by a neuritic reaction composed of axonal dystrophic processes, immunoreactive for many phosphotau epitopes and for the human tau transgenic protein. Ultrastructural observation showed in these dystrophic neurites a disorganisation of the microtubule and the neurofilament network but animals that were observed up to 18 months of age did not develop neurofibrillary tangles. These results indicate that overexpression of mutant PS1, mutant APP and of wild-type human tau were not sufficient per se to drive the formation of neurofibrillary tangles in a transgenic model. The Abeta deposits, however, were associated to marked changes in cytoskeletal organisation and in tau phosphorylation in adjacent dystrophic neurites.
The structural substrates for age-associated cognitive and motor slowing are not known, but age-related white matter changes, such as ubiquitin (UBQ)-immunoreactive granular degeneration of myelin, might contribute to this slowing. To address this hypothesis we measured immunoreactivity for UBQ and myelin basic protein (MBP) in frontal white matter of age-, sex- and postmortem interval-matched cases with no cognitive impairment (NCI; N=12), mild cognitive impairment (MCI; N=14) and Alzheimer disease (AD; N=12). There were no significant correlations between UBQ in white matter and cognitive measures, but MBP was significantly lower in AD compared with NCI and MCI. MBP correlated with overall cognition as assessed by neuropsychological summary scores, as well as with timed cognitive tests and those that reflect frontal functions. An age-related decrease in MBP immunoreactivity was detected in NCI cases (r=0.71). These results support the hypothesis that white matter pathology may contribute to age-associated decline in cognition.
We evaluated the expression of the type III intermediate filament (IF) protein, peripherin (PRP), in ubiquinated inclusions of motor neurons in amyotrophic lateral sclerosis (ALS). A previous study showed that overexpression of PRP in transgenic mice induces motor neuron disease with formation of PRP-containing inclusions before onset of symptoms [J. Cell Biol. 147 (3) (1999) 531]. To determine whether PRP inclusions occur in the human disease, we applied doublelabeling immunofluorescence to paraffin sections of the spinal cord obtained by autopsy of 40 ALS patients with sporadic disease and 39 controls. Inclusions that expressed immunoreactive ubiquitin and peripherin were recorded by video camera, and the sections were stained by hematoxylin and eosin (H&E) to define morphology. Lewy body-like inclusions (LBLIs) were seen in motor neuron perikarya of 9 of 40 ALS cases and none in controls; all LBLIs expressed peripherin. Skein-like inclusions (SLIs) were identified by ubiquitin, but did not express PRP with rare exceptions. Neither skein-like inclusions nor LBLIs expressed alpha B-crystallin, neurofilament protein (NF-L, NF-M and NF-H subunits), alpha-internexin, actin or alpha-synuclein. Immunoblot of the whole spinal cord exhibited a single 57-kDa band of peripherin in ALS patients and controls. Our data document the expression of peripherin in LBLIs, which may provide a clue to the pathogenesis of neurodegeneration in ALS.
PROTEOMEX, an approach which combines conventional proteome analysis with serological screening, is a powerful tool to separate proteins and identify immunogenic components in malignant diseases. By applying this approach, we characterized nine metabolic enzymes which were differentially expressed in renal cell carcinoma (RCC) cell lines and compared their expression profiles to that of normal kidney epithelium cells. Four of these proteins, superoxide dismutase (SODC), triosephosphatase isomerase (TPIS), thioredoxin (THIO) and ubiquitin carboxyl-terminal hydrolase (UBL1) were further analysed for both their constitutive and interferon (IFN)-gamma inducible protein expression pattern in cell lines or tissue specimens derived from RCC or normal kidney epithelium using Western blot analysis and immunohistochemistry, respectively. With the exception of the RCC cell line MZ1940RC, which completely lacks the expression of UBL1, a heterogeneous and variable expression pattern of the different metabolic enzymes was detected in RCC and normal renal epithelium. The highest differences in the expression levels were found for THIO in the RCC cell lines, which was 2-fold upregulated when compared to autologous normal kidney epithelium. Moreover, IFN-gamma treatment did not influence the constitutive expression of these metabolic enzymes. Thus, PROTEOMEX represents a valuable approach for the identification of metabolic enzymes which might be used as markers for the diagnosis of RCC.
BACKGROUND:
Stress proteins have been found to play important protective roles against ischemic brain injury under hypoxic, oxidative, heat shock, and proteasome stresses.
METHODS:
In the present study, we investigated the temporal profiles of the major stress proteins including hypoxia-inducible factor-1α (HIF-1α), glutathione (GSH), heat shock protein 72 (HSP72), constitutive heat shock cognate protein 73 (HSC73), and ubiquitin after 45 minutes of transient middle cerebral artery occlusion (tMCAO) in the mice brain up to 7 days after reperfusion.
RESULTS:
Immunohistochemical analyses of HIF-1α, GSH, HSP72, and ubiquitin showed little immunoreactivity of neural cells in sham control brain, whereas HSC73 showed a constitutive immunoreactivity. After tMCAO, HSC73 showed the fastest increase at 12 hours in the peri-ischemic area, followed by HIF-1α with a peak at 24 hours, GSH, HSP72, and ubiquitin with a peak at 72 hours. All these stress proteins returned toward the baseline levels until 7 days. In the ischemic core, these stress proteins showed a similar change with less reaction compared to the peri-ischemic area.
CONCLUSIONS:
These data showed temporal expressions of HIF-1α, GSH, HSP72, HSC73, and ubiquitin in the mice brain after tMCAO, which might provide a better understanding of neuroprotective mechanisms and novel targets for therapeutic intervention of brain ischemic disease.
Copyright © 2016 National Stroke Association. Published by Elsevier Inc. All rights reserved.
![Immunocytochemistry/Immunofluorescence - Anti-Ubiquitin Antibody [FK2] (A305218) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305218_1.png?profile=product_alternative)
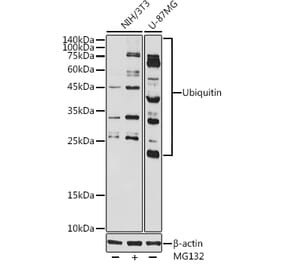
![Western Blot - Anti-Ubiquitin Antibody [ARC50024] (A307666) - Antibodies.com](https://cdn.antibodies.com/image/catalog/307/A307666_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Ubiquitin Antibody [UBB/1748] - BSA and Azide free (A253441) - Antibodies.com](https://cdn.antibodies.com/image/catalog/253/A253441_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Ubiquitin Antibody [UBB/1748] (A250261) - Antibodies.com](https://cdn.antibodies.com/image/catalog/250/A250261_1.jpg?profile=product_alternative)
![Western Blot - Anti-Ubiquitin Antibody [5B9-B3] (A305078) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305078_1.png?profile=product_alternative)
![Immunocytochemistry/Immunofluorescence - Anti-Ubiquitin Antibody [6C11-B3] (A305079) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305079_1.png?profile=product_alternative)
![Immunohistochemistry - Anti-Ubiquitin Antibody [UBB/2122] - BSA and Azide free (A253442) - Antibodies.com](https://cdn.antibodies.com/image/catalog/253/A253442_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Ubiquitin Antibody [UBB/2122] (A250262) - Antibodies.com](https://cdn.antibodies.com/image/catalog/250/A250262_1.jpg?profile=product_alternative)