

Unconjugated
The members of the epidermal growth factor receptor (EGFR) kinase family are important players in breast morphogenesis and cancer. EGFR2/HER2 and EGFR expression have a prognostic value in certain subtypes of breast cancer such as HER2-amplified, basal-like and luminal type B. Many clinically approved small molecular inhibitors and monoclonal antibodies have been designed to target HER2, EGFR or both. There is, however, still limited knowledge on how the two receptors are expressed in normal breast epithelium, what effects they have on cellular differentiation and how they participate in neoplastic transformation. D492 is a breast epithelial cell line with stem cell properties that can undergo epithelial to mesenchyme transition (EMT), generate luminal- and myoepithelial cells and form complex branching structures in three-dimensional (3D) culture. Here, we show that overexpression of HER2 in D492 (D492(HER2)) resulted in EMT, loss of contact growth inhibition and increased oncogenic potential in vivo. HER2 overexpression, furthermore, inhibited endogenous EGFR expression. Re-introducing EGFR in D492(HER2) (D492(HER2/EGFR)) partially reversed the mesenchymal state of the cells, as an epithelial phenotype reappeared both in 3D cultures and in vivo. The D492(HER2/EGFR) xenografts grow slower than the D492(HER2) tumors, while overexpression of EGFR alone (D492(EGFR)) was not oncogenic in vivo. Consistent with the EGFR-mediated epithelial phenotype, overexpression of EGFR drove the cells toward a myoepithelial phenotype in 3D culture. The effect of two clinically approved anti-HER2 and EGFR therapies, trastuzumab and cetuximab, was tested alone and in combination on D492(HER2) xenografts. While trastuzumab had a growth inhibitory effect compared with untreated control, the effect of cetuximab was limited. When administered in combination, the growth inhibitory effect of trastuzumab was less pronounced. Collectively, our data indicate that in HER2-overexpressing D492 cells, EGFR can behave as a tumor suppressor, by pushing the cells towards epithelial differentiation.
Pancreatic ductal adenocarcinoma is an unsolved health problem with nearly 75% of patients diagnosed with advanced disease and an overall 5-year survival rate near 5%. Despite the strong link between mortality and malignancy, the mechanisms behind pancreatic cancer dissemination and metastasis are poorly understood. Correlative pathological and cell culture analyses suggest the chemokine receptor CXCR4 plays a biological role in pancreatic cancer progression. In vivo roles for the CXCR4 ligand CXCL12 in pancreatic cancer malignancy were investigated. CXCR4 and CXCR7 were consistently expressed in normal and cancerous pancreatic ductal epithelium, established cell lines, and patient-derived primary cancer cells. Relative to healthy exocrine ducts, CXCL12 expression was pathologically repressed in pancreatic cancer tissue specimens and patient-derived cell lines. To test the functional consequences of CXCL12 silencing, pancreatic cancer cell lines stably expressingthe chemokine were engineered. Consistent with a role for CXCL12 as a tumor suppressor, cells producing the chemokine wereincreasingly adherent and migration deficient in vitro and poorly metastatic in vivo, compared to control cells. Further, CXCL12 reintroduction significantly reduced tumor growth in vitro, with significantly smaller tumors in vivo, leading to a pronounced survival advantage in a preclinical model. Together, these data demonstrate a functional tumor suppressive role for the normal expression of CXCL12 in pancreatic ducts, regulating both tumor growth andcellulardissemination to metastatic sites.
![Western Blot - Anti-Cytokeratin 19 Antibody [A53-B/A2] (A86354) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86355_595.jpg?profile=product_top)
![Western Blot - Anti-Cytokeratin 19 Antibody [A53-B/A2] (A86354) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86355_595.jpg?profile=product_top_thumb)
![Western Blot - Anti-Cytokeratin 19 Antibody [A53-B/A2] (A86354) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86355_595.jpg?profile=product_image)




























![Immunohistochemistry - Anti-Cytokeratin 19 Antibody [KRT19/800] (A249214) - Antibodies.com](https://cdn.antibodies.com/image/catalog/249/A249214_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Cytokeratin 19 Antibody [KRT19/800] - BSA and Azide free (A252394) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252394_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Cytokeratin 19 Antibody [KRT19/799] (A249213) - Antibodies.com](https://cdn.antibodies.com/image/catalog/249/A249213_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Cytokeratin 19 Antibody [KRT19/799] - BSA and Azide free (A252393) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252393_1.jpg?profile=product_alternative)
![Western Blot - Anti-Cytokeratin 19 Antibody [ARC0272] (A308569) - Antibodies.com](https://cdn.antibodies.com/image/catalog/308/A308569_1.jpg?profile=product_alternative)
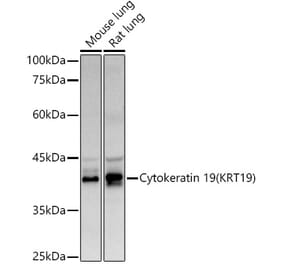
![Western Blot - Anti-Cytokeratin 19 Antibody [ARC2811] (A307943) - Antibodies.com](https://cdn.antibodies.com/image/catalog/307/A307943_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Cytokeratin 19 Antibody [A53-B/A2.26] (A249208) - Antibodies.com](https://cdn.antibodies.com/image/catalog/249/A249208_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Cytokeratin 19 Antibody [A53-B/A2.26] - BSA and Azide free (A252388) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252388_1.jpg?profile=product_alternative)
![Western Blot - Anti-Cytokeratin 19 Antibody [rKRT19/799] (A249207) - Antibodies.com](https://cdn.antibodies.com/image/catalog/249/A249207_1.jpg?profile=product_alternative)
![Western Blot - Anti-Cytokeratin 19 Antibody [rKRT19/799] - BSA and Azide free (A252387) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252387_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Cytokeratin 19 Antibody [BA17] (A249210) - Antibodies.com](https://cdn.antibodies.com/image/catalog/249/A249210_1.jpg?profile=product_alternative)