

Unconjugated
Bst-2/Tetherin inhibits the release of HIV by tethering newly formed virus particles to the plasma membrane of infected cells. Although the mechanisms of Tetherin-mediated restriction are increasingly well understood, the biological relevance of this restriction in the natural target cells of HIV is unclear. Moreover, whether Tetherin exerts any restriction on the direct cell-cell spread of HIV across intercellular contacts remains controversial. Here we analyse the restriction endogenous Tetherin imposes on HIV transmission from primary human macrophages, one of the main targets of HIV in vivo. We find that the mRNA and protein levels of Tetherin in macrophages are comparable to those in T cells from the same donors, and are highly upregulated by type I interferons. Improved immunocytochemistry protocols enable us to demonstrate that Tetherin localises to the cell surface, the trans-Golgi network, and the macrophage HIV assembly compartments. Tetherin retains budded virions in the assembly compartments, thereby impeding the release and cell-free spread of HIV, but it is not required for the maintenance of these compartments per se. Notably, using a novel assay to quantify cell-cell spread, we show that Tetherin promotes the transfer of virus clusters from macrophages to T cells and thereby restricts the direct transmission of a dual-tropic HIV-1. Kinetic analyses provide support for the notion that this direct macrophage-T cell spread is mediated, at least in part, by so-called virological synapses. Finally, we demonstrate that the viral Vpu protein efficiently downregulates the cell surface and overall levels of Tetherin, and thereby abrogates this HIV restriction in macrophages. Together, our study shows that Tetherin, one of the most potent HIV restriction factors identified to date, can inhibit virus spread from primary macrophages, regardless of the mode of transmission.
The glycosylphosphatidylinositol (GPI)-anchored molecule CD59 has been implicated in the modulation of T cell responses, but the underlying molecular mechanism of CD59 influencing T cell signaling remained unclear. Here we analyzed Jurkat T cells stimulated via anti-CD3ε- or anti-CD59-coated surfaces, using time-resolved single-cell Ca(2+) imaging as a read-out for stimulation. This analysis revealed a heterogeneous Ca(2+) response of the cell population in a stimulus-dependent manner. Further analysis of T cell receptor (TCR)/CD3 deficient or overexpressing cells showed that CD59-mediated signaling is strongly dependent on TCR/CD3 surface expression. In protein co-patterning and fluorescence recovery after photobleaching experiments no direct physical interaction was observed between CD59 and CD3 at the plasma membrane upon anti-CD59 stimulation. However, siRNA-mediated protein knock-downs of downstream signaling molecules revealed that the Src family kinase Lck and the adaptor molecule linker of activated T cells (LAT) are essential for both signaling pathways. Furthermore, flow cytometry measurements showed that knock-down of Lck accelerates CD3 re-expression at the cell surface after anti-CD59 stimulation similar to what has been observed upon direct TCR/CD3 stimulation. Finally, physically linking Lck to CD3ζ completely abolished CD59-triggered Ca(2+) signaling, while signaling was still functional upon direct TCR/CD3 stimulation. Altogether, we demonstrate that Lck mediates signal transmission from CD59 to the TCR/CD3 pathway in Jurkat T cells, and propose that CD59 may act via Lck to modulate T cell responses.
![Mass Cytometry - Anti-CD3 Antibody [MEM-57] (A86065) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86066_410.jpg?profile=product_top)
![Mass Cytometry - Anti-CD3 Antibody [MEM-57] (A86065) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86066_410.jpg?profile=product_top_thumb)
![Mass Cytometry - Anti-CD3 Antibody [UCHT1] (A86521) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86522_697.jpg?profile=product_alternative)

![Mass Cytometry - Anti-CD3 Antibody [MEM-57] (A86065) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86066_410.jpg?profile=product_image)









![Mass Cytometry - Anti-CD3 Antibody [UCHT1] - Low endotoxin, Azide free (A86528) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86529_702.jpg?profile=product_alternative)
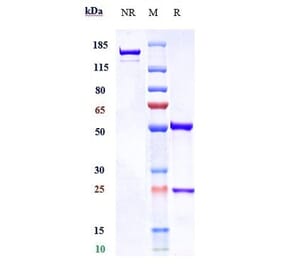
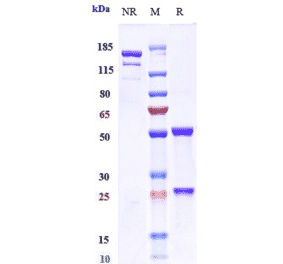
![Immunohistochemistry - Anti-CD3 Antibody [RM344] (A121367) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121411_1.png?profile=product_alternative)
![Flow Cytometry - Anti-CD3 Antibody [TB3] (A121786) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121789_1.jpg?profile=product_alternative)