

Unconjugated
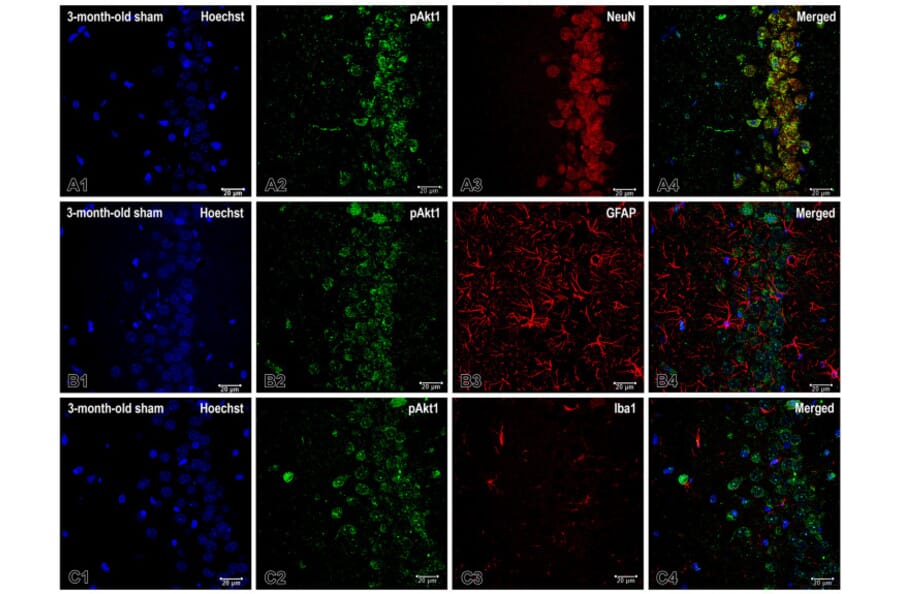
It is widely accepted that chronic inflammation constitutes a significant mechanism that promotes the biological aging process. The pineal gland is regarded as being closely related to the control of the "life clock". The present study aimed to determine the inflammation associated with pinealectomy in the rat hippocampus and to investigate the extent to which age stage impacts the severity of this inflammation. We evaluated the expression of the Akt/NF-kB signaling pathway in neurons and gliosis level in the dorsal hippocampus (dHipp) of rats subjected to sham surgery or pinealectomy at 3, 14, or 18 months of age. The assessment was conducted using immunohistochemistry. Removal of the pineal gland resulted in significant, region-specific increases in NF-kB expression in neurons of the dHipp in the youngest and middle-aged groups. However, the change in expression of the phosphorylated form of Akt (pAkt1) in neurons went in the opposite direction in these two age groups, and there were also regional differences. Pinealectomy triggered microgliosis in both young and old rats, but middle-aged rats were resistant to microglia activation. Conversely, astrogliosis was observed in young adult and middle-aged groups with melatonin deficiency in certain regions of the dHipp. It is noteworthy that young adult rats demonstrated the highest degree of vulnerability to inflammation associated with the loss of melatonin as a hormone. In contrast, middle-aged rats with pinealectomy exhibited a complex and partial adaptive response. These findings emphasize the dynamic and age-dependent nature of neuroinflammation following pinealectomy, underscoring the developmental stage as a critical determinant of inflammatory susceptibility.
![Immunofluorescence - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_1.jpg?profile=product_top)
![Immunofluorescence - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_2.jpg?profile=product_top)
![Immunofluorescence - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_3.jpg?profile=product_top)
![Immunofluorescence - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_4.jpg?profile=product_top)
![Western Blot - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_5.jpg?profile=product_top)
![Western Blot - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_6.jpg?profile=product_top)
![Binding curve - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_7.jpg?profile=product_top)
![Immunohistochemistry - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_8.jpg?profile=product_top)
![Immunofluorescence - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_1.jpg?profile=product_top_thumb)
![Immunofluorescence - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_2.jpg?profile=product_top_thumb)
![Immunofluorescence - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_3.jpg?profile=product_top_thumb)
![Immunofluorescence - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_4.jpg?profile=product_top_thumb)
![Western Blot - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_5.jpg?profile=product_top_thumb)
![Western Blot - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_6.jpg?profile=product_top_thumb)
![Binding curve - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_7.jpg?profile=product_top_thumb)
![Immunofluorescence - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_1.jpg?profile=product_image)
![Immunofluorescence - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_2.jpg?profile=product_image)
![Immunofluorescence - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_3.jpg?profile=product_image)
![Immunofluorescence - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_4.jpg?profile=product_image)
![Western Blot - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_5.jpg?profile=product_image)
![Western Blot - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_6.jpg?profile=product_image)
![Binding curve - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_7.jpg?profile=product_image)
![Immunohistochemistry - Anti-NeuN Antibody [1B7] (A85405) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85405_8.jpg?profile=product_image)


![Western Blot - Anti-NeuN Antibody [ARC0202] (A306978) - Antibodies.com](https://cdn.antibodies.com/image/catalog/306/A306978_1.jpg?profile=product_alternative)
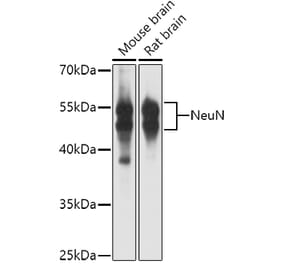
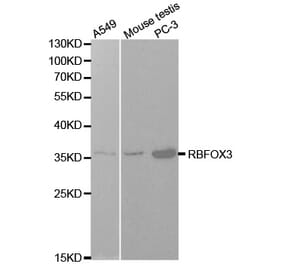
![Immunohistochemistry - Anti-NeuN Antibody [NeuN/288R] (A277965) - Antibodies.com](https://cdn.antibodies.com/image/catalog/277/A277965_1.jpg?profile=product_alternative)
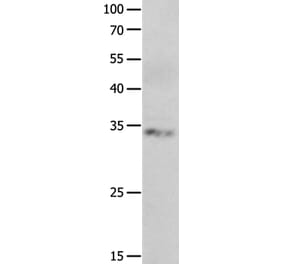
![Immunohistochemistry - Anti-NeuN Antibody [NeuN/288R] - BSA and Azide free (A278553) - Antibodies.com](https://cdn.antibodies.com/image/catalog/278/A278553_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-NeuN Antibody [NeuN/6694R] - BSA and Azide free (A278554) - Antibodies.com](https://cdn.antibodies.com/image/catalog/278/A278554_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-NeuN Antibody [NeuN/6694R] (A277966) - Antibodies.com](https://cdn.antibodies.com/image/catalog/277/A277966_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-NeuN Antibody [NeuN/7071R] (A277967) - Antibodies.com](https://cdn.antibodies.com/image/catalog/277/A277967_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-NeuN Antibody [RM312] (A121402) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121385_1.png?profile=product_alternative)