Unconjugated
Rapid induction of transcription is known to be mediated by factors which bind DNA following post-translational modification. We report here that non-tyrosine phosphorylated (NTP)-Stat1 is involved in a cooperative interaction with Spi-1/PU.1 and IRF8 to form a pre-associated, poised complex for IL1B gene induction. A double point mutation at a putative STAT binding site, which overlaps this composite Spi-1 x IRF8 site located in the LPS and IL-1 response element (LILRE), inhibited human IL1B LPS-dependent reporter activity to about 10 percent of the control wild type vector. Chromatin immunoprecipitation revealed stimulation-independent constitutive binding of IRF8, Spi-1 and NTP-Stat1 at the LILRE, while binding of C/EBP beta was activated at an adjacent C/EBP beta site after LPS stimulation. In contrast to Stat1, IRF8 was tyrosine phosphorylated following LPS treatment. Supporting the involvement of NTP-Stat1, LPS-induced IL1B reporter activity in monocytes was enhanced by ectopic expression of NTP-Stat1 Y701F. In contrast, co-expression of a Y211F IRF8 mutein functioned as a dominant-negative inhibitor of LPS-induced IL1B reporter activity. In vitro DNA binding using extracts from LPS-treated monocytes confirmed that the LILRE enhancer constitutively binds a trimolecular complex containing IRF8, Spi-1 and NTP-Stat1. Binding studies using in vitro-expressed proteins revealed that NTP-Stat1 enhanced the binding of Spi-1 and IRF8 to the LILRE. Co-expression of TRAF6, an LPS surrogate, with Spi-1 and IRF8 enhanced IL1B reporter activity in HEK293R cells, which was dramatically reduced when Y211F IRF8 was co-expressed. These results suggest that the rapid transcriptional induction of an important inflammatory gene is dependent upon constitutive cooperative binding of a Spi-1 x IRF8 x NTP-Stat1 complex to the LILRE, which primes the gene for immediate induction following IRF8 phosphorylation. Phosphorylation of chromatin pre-associated factors like IRF8 may be an important strategy for the rapid transcriptional activation of genes involved in innate immunity.
The 18-kDa TSPO (translocator protein) localizes on the outer mitochondrial membrane (OMM) and participates in cholesterol transport. Here, we report that TSPO inhibits mitochondrial autophagy downstream of the PINK1-PARK2 pathway, preventing essential ubiquitination of proteins. TSPO abolishes mitochondrial relocation of SQSTM1/p62 (sequestosome 1), and consequently that of the autophagic marker LC3 (microtubule-associated protein 1 light chain 3), thus leading to an accumulation of dysfunctional mitochondria, altering the appearance of the network. Independent of cholesterol regulation, the modulation of mitophagy by TSPO is instead dependent on VDAC1 (voltage-dependent anion channel 1), to which TSPO binds, reducing mitochondrial coupling and promoting an overproduction of reactive oxygen species (ROS) that counteracts PARK2-mediated ubiquitination of proteins. These data identify TSPO as a novel element in the regulation of mitochondrial quality control by autophagy, and demonstrate the importance for cell homeostasis of its expression ratio with VDAC1.






![Flow Cytometry - Anti-STAT1 Antibody [SM1] (A85869) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85870_279.jpg?profile=product_alternative)
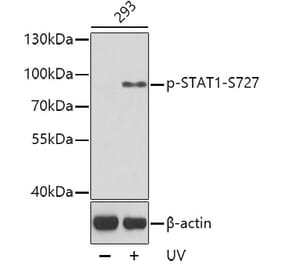
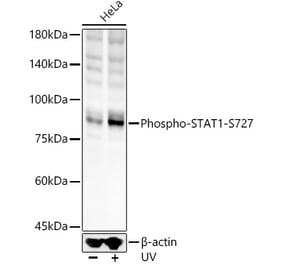
![Western Blot - Anti-STAT1 (phospho Ser727) Antibody [ARC1544] (A308199) - Antibodies.com](https://cdn.antibodies.com/image/catalog/308/A308199_1.jpg?profile=product_alternative)
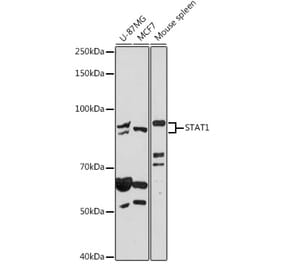
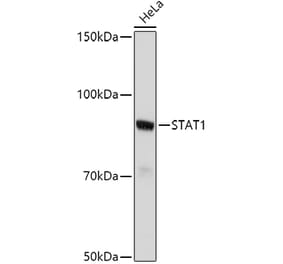
![Western Blot - Anti-STAT1 Antibody [ARC0042] (A307829) - Antibodies.com](https://cdn.antibodies.com/image/catalog/307/A307829_1.jpg?profile=product_alternative)
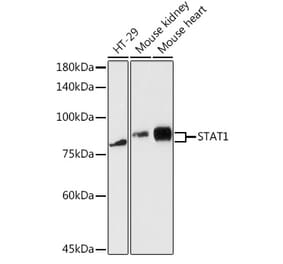

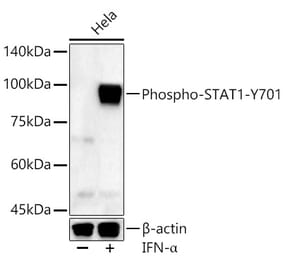
![Western Blot - Anti-STAT1 (phospho Tyr701) Antibody [ARC0049] (A308896) - Antibodies.com](https://cdn.antibodies.com/image/catalog/308/A308896_1.jpg?profile=product_alternative)