Unconjugated
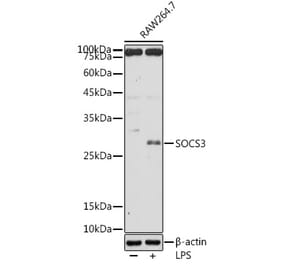
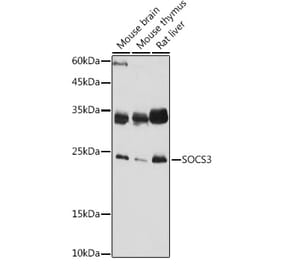
The main pathogenesis of saphenous vein graft neointimal hyperplasia after coronary artery bypass grafting (CABG) is inflammation-caused migration and proliferation of vascular smooth muscle cells (VSMCs). Janus kinase 2/signal transducer and activators of transcription 3 (JAK2/STAT3) pathway is an important signaling pathway through which VSMCs phenotype conversion occurs. Suppressor of cytokine signaling 3 (SOCS3) is the classic negative feedback inhibitor of JAK2/STAT3 pathway. Growing studies show that SOCS3 plays an important anti-inflammatory role in numerous autoimmune diseases, inflammatory diseases and inflammation-related tumors. However, the effect and mechanism of SOCS3 on vein graft disease is unclear. The purpose of this study was to investigate the effects of SOCS3 on the inflammation, migration and proliferation of VSMCs in vitro and the mechanism. The small interference RNA plasmid targeting rat SOCS3 (SiRNA-rSOCS3) and the recombinant adenovirus vector carrying rat SOCS3 gene (pYrAd-rSOCS3) were constructed, and the empty plamid (SiRNA-control) and vector (pYrAd-GFP) only carrying GFP reported gene were constructed as control. The rat VSMCs were cultured. There were two large groups of A (SOCS3 up-regulated): control group, IL-6/IFN-γ group, IL-6/IFN-γ+pYrAd-rSOCS3 group, IL-6/IFN-γ(+)pYrAd-GFP group; and B (SOCS3 down-regulated): control group, IL-6/IFN-γ group, IL-6/IFN-γ+SiRNA-rSOCS3 group and IL-6/ IFN -γ+SiRNA-control group. The pYrAd-rSOCS3 and SiRNA-rSOCS3 were transfected into VSMCs induced by IL-6/IFN-γ. After 24 h, real-time reverse transcription polymerase chain reaction (RT-PCR) and Western blotting were used to detect the mRNA and protein expression of SOCS3, STAT3 (only by Western blotting), P-STAT3 (only by Western blotting), IL-1β, IL-6, TNF-α, MCP-1 and ICAM-1. The MTT, Transwell assay and flow cytometry were used to examine VSMCs proliferation, migration and cell cycle progression, respectively. As compared with control group, the mRNA and protein expression of SOCS3, STAT3, P-STAT3, IL-1β, IL-6, TNF-α, MCP-1 and ICAM-1 was significantly up-regulated in VSMCs stimulated by IL-6/IFN-γ. However, in VSMCs transfected with pYrAd-rSOCS3 before stimulation with IL-6/IFN-γ, the expression of SOCS3 mRNA and protein was further up-regulated, and that of STAT3, P-STAT3, IL-1β, IL-6, TNF-α, MCP-1 and ICAM-1 was significantly down-regulated as compared with IL-6/IFN-γ group and IL-6/IFN-γ+pYrAd-GFP group. The expression of those related-cytokines in IL-6/IFN-γ+SiRNA-rSOCS3 group was markedly increased as compared with IL-6/IFN-γ group and IL-6/IFN-γ+SiRNA-control group. The absorbance (A) values, the number of cells migrating to the lower chamber, and percentage of cells in the G2/M+S phase were increased in VSMCs stimulated by IL-6/IFN-γ. In VSMCs incubated with pYrAd-rSOCS3 or SiRNA-rSOCS3 before IL-6/IFN-γ stimulation, the A values, the number of cells migrating to the lower chamber, and the percentage of cells in the G2/M+S phase were significantly decreased, and increased respectively. These results imply that IL-6/IFN-γ, strong inflammatory stimulators, can promote transformation of VSMCs phenotype form a quiescent contractile state to a synthetic state by activating JAK2/STAT3 pathway. Over-expresssed SOCS3 might inhibit pro-inflammatory effect, migration and growth of VSMCs by blocking STAT3 activation and phosphorylation. These data in vitro confirm that SOCS3 may play a negatively regulatory role in development and progression of vein graft failure. These conclusions can provide a novel strategy for clinical treatment of vein graft diseases and a new theoretic clue for related drug development.
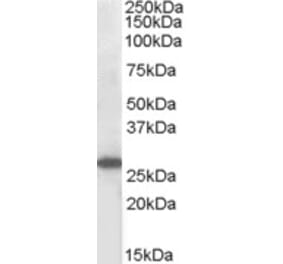
Suppressors of cytokine signalling (SOCS) are induced by interleukins (ILs) and various peptide hormones and may prevent sustained activation of signalling pathways. We have previously shown that SOCS-3 antagonizes regulation of cellular events by cAMP and is expressed in human prostate cancer. To investigate possible effects of androgen on SOCS-3 protein expression, two prostate cancer cell lines (PC3-AR and LAPC4) were treated with different concentrations of R1881. Western blot analyses revealed induction of SOCS-3 protein expression in both cell lines by androgen, an effect which can be blocked by the anti-androgen bicalutamide. To further characterize the effects of R1881 on the SOCS-3 gene, promoter-reporter assay and real-time PCR were performed. We found no influence of androgen on promoter activity or SOCS-3 mRNA levels, thus suggesting a post-transcriptional effect of androgen. Concordant with our previous findings, we show a significant increase of SOCS-3 protein after androgen treatment in cells in which transcription was blocked, but not in those with impaired translation. In order to understand implications of SOCS-3 regulation by androgen, we used SOCS-3-negative LNCaP-IL-6 cells and stably transfected them with a tetracycline-responsive SOCS-3 Tet-On plasmid. We report that androgenic effects on cell proliferation and prostate-specific antigen secretion are significantly diminished following up-regulation of SOCS-3. In conclusion, androgen up-regulates SOCS-3 protein via post-transcriptional effects. SOCS-3 inhibits androgen-stimulated proliferation by influencing cell cycle regulation. Taken together with previous findings showing androgen receptor activation by IL-6, our results imply that androgen and cytokine signalling pathways interact at multiple levels in prostate cancer.
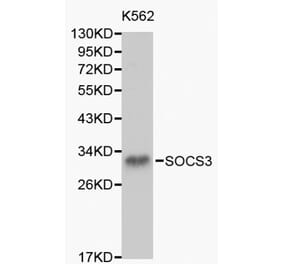
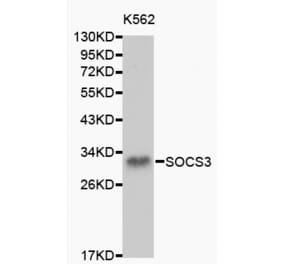
![Western Blot - Anti-SOCS3 Antibody [SO1] (A86786) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86787_894.jpg?profile=product_top)
![Western Blot - Anti-SOCS3 Antibody [SO1] (A86786) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86787_894.jpg?profile=product_top_thumb)
![Western Blot - Anti-SOCS3 Antibody [SO1] (A86786) - Antibodies.com](https://cdn.antibodies.com/image/catalog/86/A86787_894.jpg?profile=product_image)



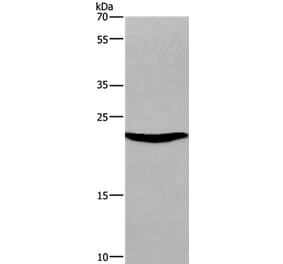
![Western Blot - Anti-SOCS3 Antibody [ARC53312] (A306254) - Antibodies.com](https://cdn.antibodies.com/image/catalog/306/A306254_1.jpg?profile=product_alternative)