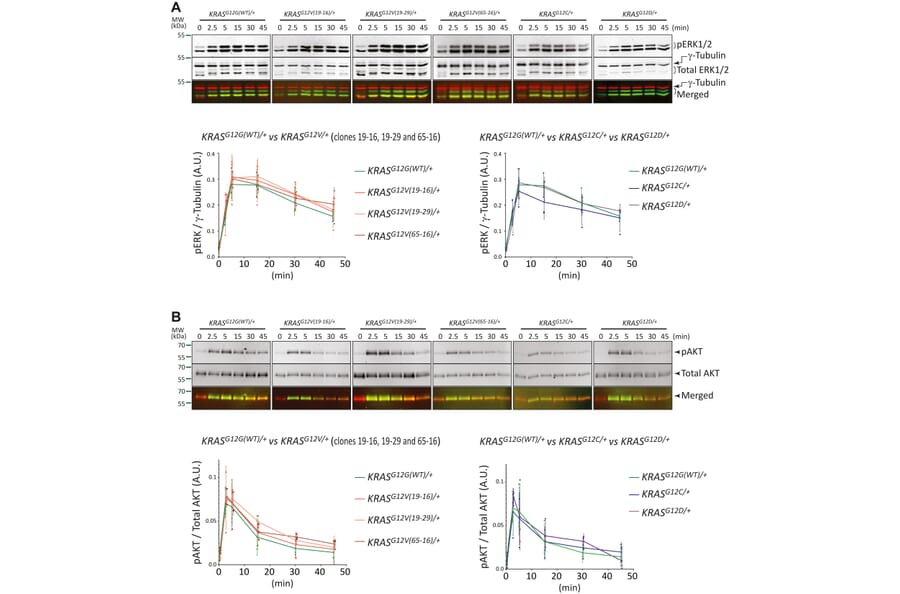
About 18% of all human cancers carry a mutation in the KRAS gene making it among the most sought-after anticancer targets. However, mutant KRas protein has proved remarkably undruggable. The recent approval of the first generation of RAS inhibitors therefore marks a seminal milestone in the history of cancer research. It also raises the predictable challenges of limited drug efficacies and acquired resistance. Hence, new approaches that improve our understanding of the tumorigenic mechanisms of oncogenic RAS within more physiological settings continue to be essential. Here, we have used the near-diploid hTERT RPE-1 cells to generate isogenic cell lines in which one of the endogenous KRAS alleles carries an oncogenic KRAS mutation at glycine 12. Cells with a KRASG12V/+, KRASG12C/+, or KRASG12D/+ genotype, together with WT KRASG12G(WT)/+ cells, reveal that oncogenic KRAS.G12X mutations increase cell proliferation rate and cell motility and reduced focal adhesions in KRASG12V/+ cells. Epidermal growth factor -induced phosphorylation of ERK and AKT was comparable between KRASG12V/+, KRASG12C/+, KRASG12D/+, and KRASG12G(WT)/+ cells. Interestingly, KRASG12X/+ cells showed varying responses to distinct inhibitors with the KRASG12V/+ and KRASG12D/+ cells more sensitive to hydroxyurea and MEK inhibitors, U0126 and trametinib, but more resistant to PI3K inhibitor, PIK-90, than the KRASG12G(WT)/+ cells. A combination of low doses of hydroxyurea and U0126 showed an additive inhibition on growth rate that was greater in KRASG12V/+ than WT cells. Collectively, these cell lines will be a valuable resource for studying oncogenic RAS signaling and developing effective anti-KRAS reagents with minimum cytotoxicity on WT cells.