Unconjugated
Most colon cancer cases are initiated by truncating mutations in the tumor suppressor, adenomatous polyposis coli (APC). APC is a critical negative regulator of the Wnt signaling pathway that participates in a multi-protein "destruction complex" to target the key effector protein ß-catenin for ubiquitin-mediated proteolysis. Prior work has established that the poly(ADP-ribose) polymerase (PARP) enzyme Tankyrase (TNKS) antagonizes destruction complex activity by promoting degradation of the scaffold protein Axin, and recent work suggests that TNKS inhibition is a promising cancer therapy. We performed a yeast two-hybrid (Y2H) screen and uncovered TNKS as a putative binding partner of Drosophila APC2, suggesting that TNKS may play multiple roles in destruction complex regulation. We find that TNKS binds a C-terminal RPQPSG motif in Drosophila APC2, and that this motif is conserved in human APC2, but not human APC1. In addition, we find that APC2 can recruit TNKS into the ß-catenin destruction complex, placing the APC2/TNKS interaction at the correct intracellular location to regulate ß-catenin proteolysis. We further show that TNKS directly PARylates both Drosophila Axin and APC2, but that PARylation does not globally regulate APC2 protein levels as it does for Axin. Moreover, TNKS inhibition in colon cancer cells decreases ß-catenin signaling, which we find cannot be explained solely through Axin stabilization. Instead, our findings suggest that TNKS regulates destruction complex activity at the level of both Axin and APC2, providing further mechanistic insight into TNKS inhibition as a potential Wnt pathway cancer therapy.
The Wnt pathway is a conserved signal transduction pathway that contributes to normal development and adult homeostasis, but is also misregulated in human diseases such as cancer. The tumor suppressor adenomatous polyposis coli (APC) is an essential negative regulator of Wnt signaling inactivated in >80% of colorectal cancers. APC participates in a multiprotein "destruction complex" that targets the proto-oncogene ß-catenin for ubiquitin-mediated proteolysis; however, the mechanistic role of APC in the destruction complex remains unknown. Several models of APC function have recently been proposed, many of which have emphasized the importance of phosphorylation of high-affinity ß-catenin-binding sites [20-amino-acid repeats (20Rs)] on APC. Here we test these models by generating a Drosophila APC2 mutant lacking all ß-catenin-binding 20Rs and performing functional studies in human colon cancer cell lines and Drosophila embryos. Our results are inconsistent with current models, as we find that ß-catenin binding to the 20Rs of APC is not required for destruction complex activity. In addition, we generate an APC2 mutant lacking all ß-catenin-binding sites (including the 15Rs) and find that a direct ß-catenin/APC interaction is also not essential for ß-catenin destruction, although it increases destruction complex efficiency in certain developmental contexts. Overall, our findings support a model whereby ß-catenin-binding sites on APC do not provide a critical mechanistic function per se, but rather dock ß-catenin in the destruction complex to increase the efficiency of ß-catenin destruction. Furthermore, in Drosophila embryos expressing some APC2 mutant transgenes we observe a separation of ß-catenin destruction and Wg/Wnt signaling outputs and suggest that cytoplasmic retention of ß-catenin likely accounts for this difference.
![Immunohistochemistry - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_1.jpg?profile=product_top)
![Immunohistochemistry - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_2.jpg?profile=product_top)
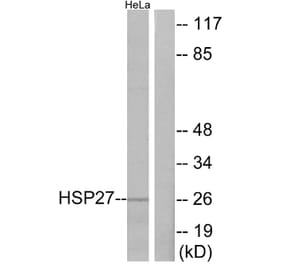
![Western Blot - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_3.jpg?profile=product_top)
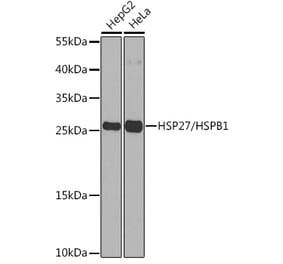
![SDS-PAGE - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_4.jpg?profile=product_top)
![Immunofluorescence - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_5.jpg?profile=product_top)
![Flow Cytometry - Anti-HSP27 Antibody [G3.1] (A248871) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_6.jpg?profile=product_top)
![Immunohistochemistry - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_1.jpg?profile=product_top_thumb)
![Immunohistochemistry - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_2.jpg?profile=product_top_thumb)
![Western Blot - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_3.jpg?profile=product_top_thumb)
![SDS-PAGE - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_4.jpg?profile=product_top_thumb)
![Immunofluorescence - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_5.jpg?profile=product_top_thumb)
![Flow Cytometry - Anti-HSP27 Antibody [G3.1] (A248871) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_6.jpg?profile=product_top_thumb)
![Immunohistochemistry - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_1.jpg?profile=product_image)
![Immunohistochemistry - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_2.jpg?profile=product_image)
![Western Blot - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_3.jpg?profile=product_image)
![SDS-PAGE - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_4.jpg?profile=product_image)
![Immunofluorescence - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_5.jpg?profile=product_image)
![Flow Cytometry - Anti-HSP27 Antibody [G3.1] (A248871) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_6.jpg?profile=product_image)

![Immunohistochemistry - Anti-HSP27 Antibody [8A7] (A304709) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304709_1.png?profile=product_alternative)
![Immunohistochemistry - Anti-HSP27 Antibody [G3.1] - BSA and Azide free (A252052) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252052_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-HSP27 Antibody [SPM252] (A248873) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248873_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-HSP27 Antibody [SPM252] - BSA and Azide free (A252053) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252053_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-HSP27 Antibody [HSPB1/774] (A248874) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248874_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-HSP27 Antibody [HSPB1/774] - BSA and Azide free (A252054) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252054_1.jpg?profile=product_alternative)
![SDS-PAGE - Anti-HSP27 Antibody [CPTC-HSPB1-2] (A248875) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248875_1.jpg?profile=product_alternative)
![Western Blot - Anti-HSP27 Antibody [5D12-A12] (A304734) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304734_1.png?profile=product_alternative)
![SDS-PAGE - Anti-HSP27 Antibody [CPTC-HSPB1-2] - BSA and Azide free (A252055) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252055_1.jpg?profile=product_alternative)