Unconjugated
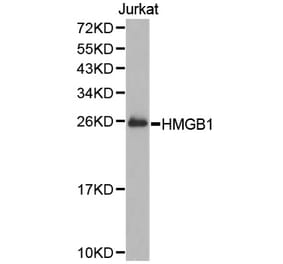
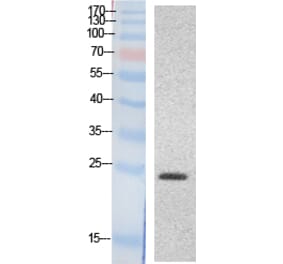
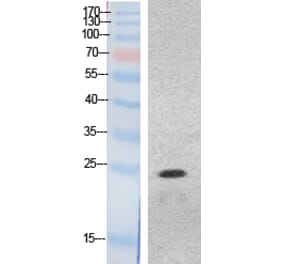
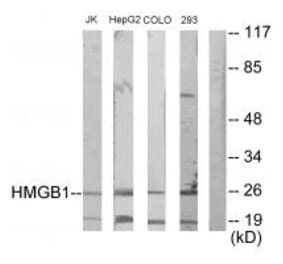
Myocardial inflammation contributes to cardiomyopathy in diabetic patients through incompletely defined underlying mechanisms. In both human and time-course experimental samples, diabetic hearts exhibited abnormal ER, with a maladaptive shift over time in rodents. Furthermore, as a cardiac ER dysfunction model, mice with cardiac-specific p21-activated kinase 2 (PAK2) deletion exhibited heightened myocardial inflammatory response in diabetes. Mechanistically, maladaptive ER stress-induced CCAAT/enhancer-binding protein homologous protein (CHOP) is a novel transcriptional regulator of cardiac high-mobility group box-1 (HMGB1). Cardiac stress-induced release of HMGB1 facilitates M1 macrophage polarization, aggravating myocardial inflammation. Therapeutically, sequestering the extracellular HMGB1 using glycyrrhizin conferred cardioprotection through its anti-inflammatory action. Our findings also indicated that an intact cardiac ER function and protective effects of the antidiabetic drug interdependently attenuated the cardiac inflammation-induced dysfunction. Collectively, we introduce an ER stress-mediated cardiomyocyte-macrophage link, altering the macrophage response, thereby providing insight into therapeutic prospects for diabetes-associated cardiac dysfunction.
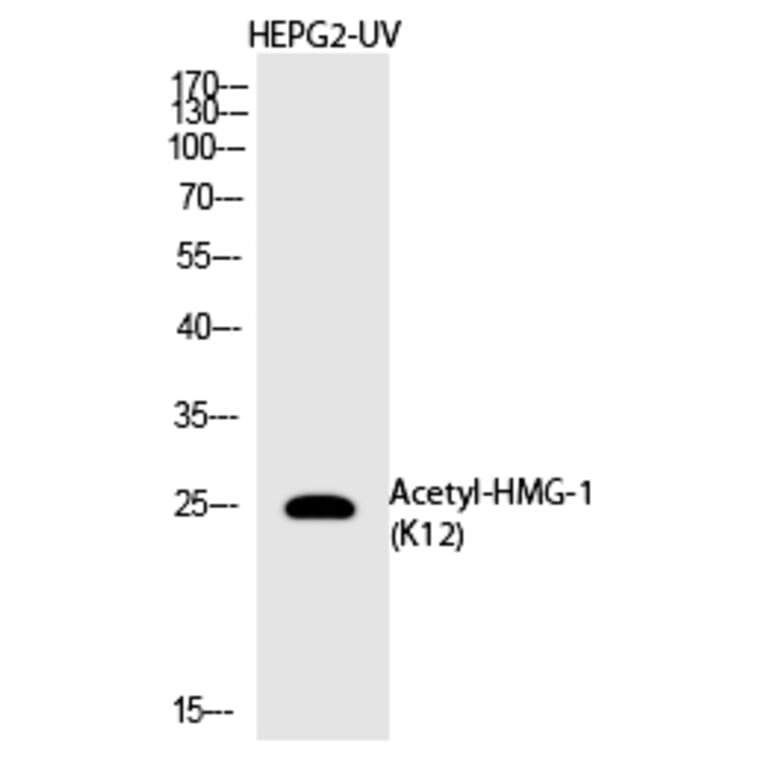
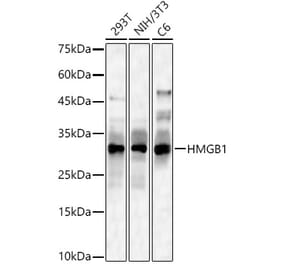
![Immunofluorescence - Anti-HMGB1 Antibody [1F3] (A85355) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85355_1.jpg?profile=product_alternative)
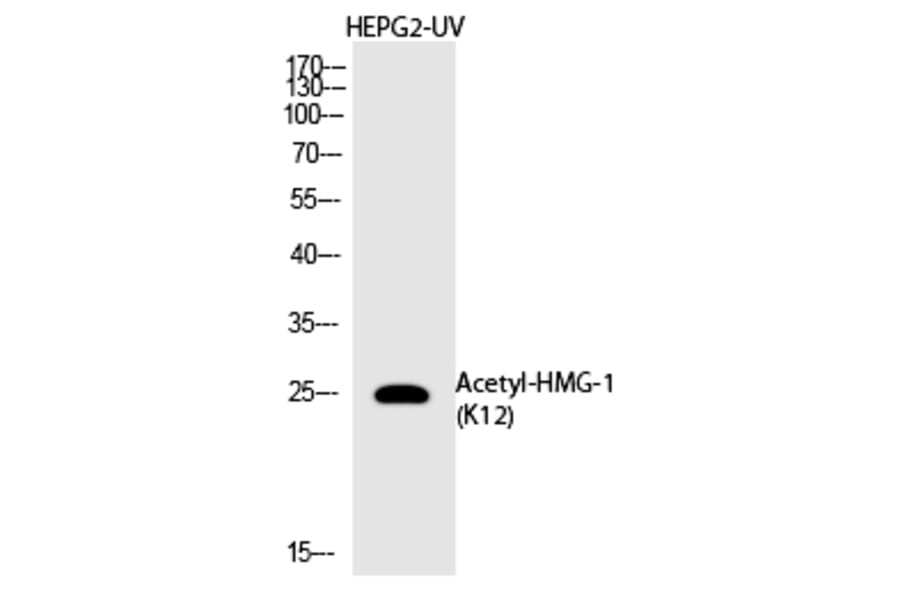


![Western Blot - Anti-HMGB1 Antibody [ARC0001] (A308594) - Antibodies.com](https://cdn.antibodies.com/image/catalog/308/A308594_1.jpg?profile=product_alternative)