Unconjugated
Adar null mutant mouse embryos die with aberrant double-stranded RNA (dsRNA)-driven interferon induction, and Adar Mavs double mutants, in which interferon induction is prevented, die soon after birth. Protein kinase R (Pkr) is aberrantly activated in Adar Mavs mouse pup intestines before death, intestinal crypt cells die, and intestinal villi are lost. Adar Mavs Eifak2 (Pkr) triple mutant mice rescue all defects and have long-term survival. Adenosine deaminase acting on RNA 1 (ADAR1) and PKR co-immunoprecipitate from cells, suggesting PKR inhibition by direct interaction. AlphaFold studies on an inhibitory PKR dsRNA binding domain (dsRBD)-kinase domain interaction before dsRNA binding and on an inhibitory ADAR1 dsRBD3-PKR kinase domain interaction on dsRNA provide a testable model of the inhibition. Wild-type or editing-inactive human ADAR1 expressed in A549 cells inhibits activation of endogenous PKR. ADAR1 dsRNA binding is required for, but is not sufficient for, PKR inhibition. Mutating the ADAR1 dsRBD3-PKR contact prevents co-immunoprecipitation, ADAR1 inhibition of PKR activity, and co-localization of ADAR1 and PKR in cells.
Quantifying differences in mRNA abundance is a classic approach to understand the impact of a given gene mutation on cell physiology. However, characterizing differences in the translatome (the whole of translated mRNAs) provides an additional layer of information particularly useful when trying to understand the function of RNA regulating or binding proteins. A number of methods for accomplishing this have been developed, including ribosome profiling and polysome analysis. However, both methods carry significant technical challenges and cannot be applied to specific cell populations within a tissue unless combined with additional sorting methods. In contrast, the RiboTag method is a genetic-based, efficient, and technically straightforward alternative that allows the identification of ribosome associated RNAs from specific cell populations without added sorting steps, provided an applicable cell-specific Cre driver is available. This method consists of breeding to generate the genetic models, sample collection, immunoprecipitation, and downstream RNA analyses. Here, we outline this process in adult male mouse germ cells mutant for an RNA binding protein required for male fertility. Special attention is paid to considerations for breeding with a focus on efficient colony management and the generation of correct genetic backgrounds and immunoprecipitation in order to reduce background and optimize output. Discussion of troubleshooting options, additional confirmatory experiments, and potential downstream applications is also included. The presented genetic tools and molecular protocols represent a powerful way to describe the ribosome-associated RNAs of specific cell populations in complex tissues or in systems with aberrant mRNA storage and translation with the goal of informing on the molecular drivers of mutant phenotypes.
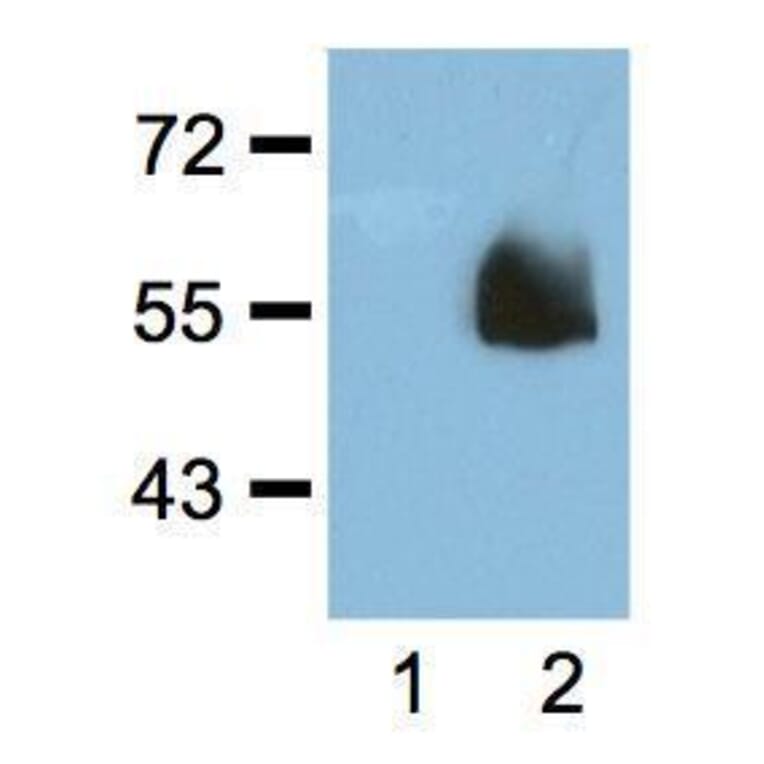
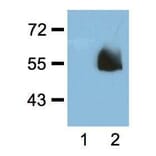
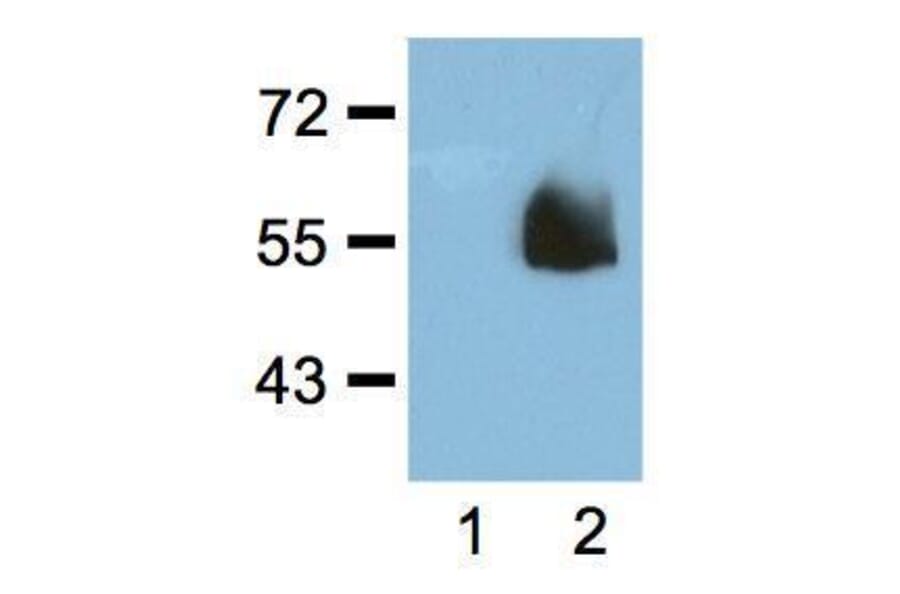

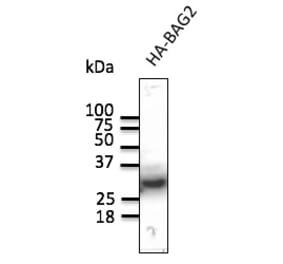
![SDS-PAGE - Anti-HA Tag Antibody [HA/279] - BSA and Azide free (A278453) - Antibodies.com](https://cdn.antibodies.com/image/catalog/278/A278453_1.jpg?profile=product_alternative)
![Western Blot - Anti-HA Tag Antibody [16.43] - BSA and Azide free (A254042) - Antibodies.com](https://cdn.antibodies.com/image/catalog/254/A254043_1.jpg?profile=product_alternative)
![Western Blot - Anti-HA Tag Antibody [RM305] (A121321) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121321_1.png?profile=product_alternative)
![Western Blot - Anti-HA Tag Antibody [16.43] (A250862) - Antibodies.com](https://cdn.antibodies.com/image/catalog/250/A250863_1.jpg?profile=product_alternative)
![SDS-PAGE - Anti-HA Tag Antibody [HA/279] (A277865) - Antibodies.com](https://cdn.antibodies.com/image/catalog/277/A277865_1.jpg?profile=product_alternative)
![ELISA - Anti-HA Tag Antibody [RMH02] (A121359) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121322_1.png?profile=product_alternative)