Unconjugated
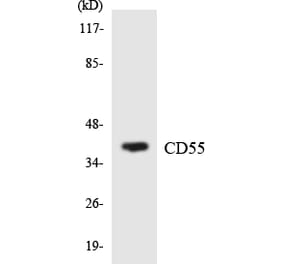
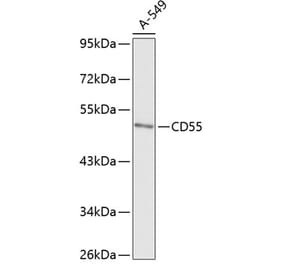
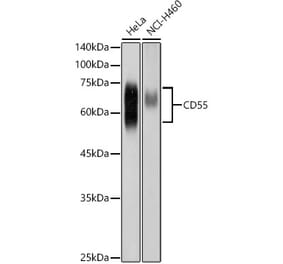
The aim of the present study was to evaluate the role of interleukin (IL)-6 and IL-8 on the expression of the membrane-bound complement inhibitors membrane attack complex-inhibitory protein (CD59) and decay-accelerating factor (CD55), in the human ovarian carcinoma A2780 cell line, which is a non-producing IL-6 cell line that does exhibit IL-6 responsiveness, due to the presence of IL-6 receptors. Extracellular levels of complement system inhibitors were evaluated by western blotting and reverse transcription-quantitative polymerase chain reaction. Cellular localization of CD55 and CD59 in the ovarian cancer cells was assessed by immunofluorescence. The detection of a soluble form of CD55 and CD59 released by the A2780 cells following stimulation with IL-6 and IL-8 was detected by enzyme-linked immunosorbent assay. The present data revealed that A2780 cells express CD55 and CD59 at the mRNA and protein level, but do not secrete these proteins to the culture medium. Results of western blotting demonstrated that the protein level of CD59 was regulated by IL-6 and IL-8 in a dose-dependent manner. Immunofluorescence analysis revealed that the ovarian cancer A2780 cell line expresses the membrane bound form of CD55 protein. The present results indicate that CD55 and CD59 may affect the efficiency of complement-mediated immunotherapies.
During evolution, herpesviruses have developed numerous, and often very ingenious, strategies to counteract efficient host immunity. Specifically, Kaposi's sarcoma-associated herpesvirus (KSHV) eludes host immunity by undergoing a dormant stage, called latency wherein it expresses a minimal number of viral proteins to evade host immune activation. Here, we show that during latency, KSHV hijacks the complement pathway to promote cell survival. We detected strong deposition of complement membrane attack complex C5b-9 and the complement component C3 activated product C3b on Kaposi's sarcoma spindle tumor cells, and on human endothelial cells latently infected by KSHV, TIME-KSHV and TIVE-LTC, but not on their respective uninfected control cells, TIME and TIVE. We further showed that complement activation in latently KSHV-infected cells was mediated by the alternative complement pathway through down-regulation of cell surface complement regulatory proteins CD55 and CD59. Interestingly, complement activation caused minimal cell death but promoted the survival of latently KSHV-infected cells grown in medium depleted of growth factors. We found that complement activation increased STAT3 tyrosine phosphorylation (Y705) of KSHV-infected cells, which was required for the enhanced cell survival. Furthermore, overexpression of either CD55 or CD59 in latently KSHV-infected cells was sufficient to inhibit complement activation, prevent STAT3 Y705 phosphorylation and abolish the enhanced survival of cells cultured in growth factor-depleted condition. Together, these results demonstrate a novel mechanism by which an oncogenic virus subverts and exploits the host innate immune system to promote viral persistent infection.








![Immunofluorescence - Anti-CD55 Antibody [143-30] - BSA and Azide free (A251499) - Antibodies.com](https://cdn.antibodies.com/image/catalog/251/A251499_1.jpg?profile=product_alternative)
![Immunofluorescence - Anti-CD55 Antibody [F4-29D9] - BSA and Azide free (A251498) - Antibodies.com](https://cdn.antibodies.com/image/catalog/251/A251498_1.jpg?profile=product_alternative)
![Immunofluorescence - Anti-CD55 Antibody [143-30] (A248317) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248317_1.jpg?profile=product_alternative)
![Immunofluorescence - Anti-CD55 Antibody [F4-29D9] (A248316) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248316_1.jpg?profile=product_alternative)

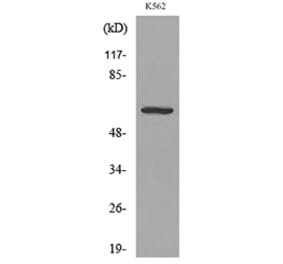
![Western Blot - Anti-CD55 Antibody [ARC0568] (A305715) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305715_1.jpg?profile=product_alternative)