Christmas disease, also known as Haemophilia B or Factor IX Deficiency, is a rare genetic bleeding disorder that results in defective blood clotting. The name ‘Christmas Disease’ originates from Stephen Christmas; who in 1952 became the first patient to be reported with this disorder in the medical literature.
Individuals with this disorder have a mutation in the F9 gene on the X chromosome, reducing expression of the clotting Factor IX 1. The severity of the disease depends on the expression levels of Factor IX; with mild classified as Factor XI levels at 5-40% of normal levels, moderate classified as Factor XI levels at 1-5% of normal levels, and severe classified as Factor XI levels at < 1% of normal levels. Christmas disease is estimated to occur in 1 in 25,000 male births, which is less prevalent than Haemophilia A, which occurs in 1 in 5,000 male births. Females are predominantly carriers of the mutant gene and do not show symptoms at the same rate, due to the disease being an X-linked recessive disorder, however, it is still estimated that 10-25% of female carriers will develop mild symptoms.
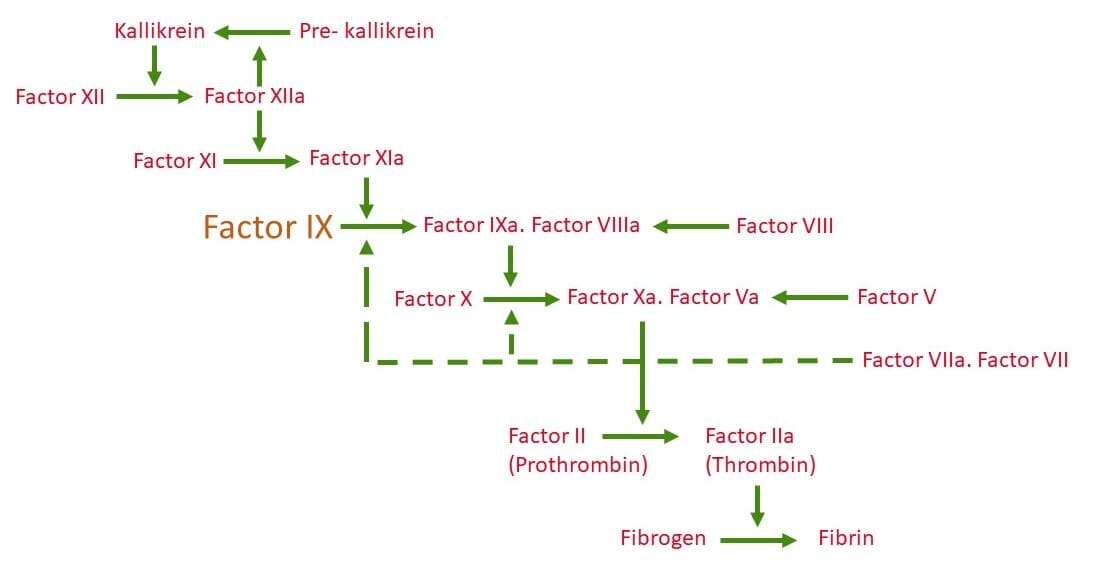
The genetic disorder is caused by an X-linked recessive genetic mutation (specifically at location Xq27. 1-q27-2) which manifests in a deficiency of clotting Factor IX [1]. The deficiency of Factor IX compromises the activation of Factor X which inhibits sufficient production of Thrombin [2], which in turn inhibits sufficient production of Fibrin (the major component of blood clots), and results in haemophilia [3].
Figure 1. Schematic of the coagulation cascade which results in the formation of Fibrin; the structural polymer of blood clots. Factor IX (shown in orange) is essential for the intrinsic pathway and a deficiency or dysfunction compromises the activation of Factor X and subsequently the production of Fibrin resulting in haemophilia [3].
Inadequate coagulation manifests into many life-threatening and distressing symptoms, including: bleeding episodes resulting in long-term disability and severe arthropathy, muscle atrophy, prolonged bleeding after an injury or medical procedure, gastrointestinal or urinary tract bleeding [4], and early death [5, 6]. However, due to recent scientific advances, the life expectancies for patients with Christmas disease have increased dramatically up until a normal life expectancy - which is a monumental success!
This feat was achieved through advances in diagnostic techniques, including activated partial thromboplastin time test, fibrinogen test, prothrombin time test, and mutant gene screening [7], which enable accurate determination of Factor IX levels, and Factor IX injections to prevent or stop bleeding [8], however, these injections are associated with peak and trough concentrations; which leaves the patient at risk of bleeding if the concentration falls too low. New research has also resulted in cost-effective continuous infusion methods which have proven successful, however, outside of clotting factor replacements there are no other treatment options currently available [9]. One research area being explored is focused on preventing or slowing arthropathy, a debilitating phenotype of Haemophilia B, which may result in additional treatment options in the future.
Biomarkers are useful targets to objectively measure and evaluate diseases for therapeutic intervention [10], and analysis of biomarkers might identify individuals for targeted treatment earlier and more efficiently.
The following biomarkers have been identified for arthropathy, specifically for cartilage degradation:
The following biomarkers have been found at increased levels in haemophilia patients: