Chromatin Immunoprecipitation ChIP
The complete ChIP technique guide.
By Ryan Hamnett, PhD
Chromatin immunoprecipitation (ChIP) is a technique for isolating chromatin that is bound to a protein of interest for subsequent identification and characterization.
Chromatin refers to the complex of DNA and protein that allows the genetic material of the cell to be packaged into the nucleus. The primary protein component of chromatin, termed histones, along with other regulatory proteins and transcription factors, interact with DNA to maintain its 3D structure and regulate gene expression.
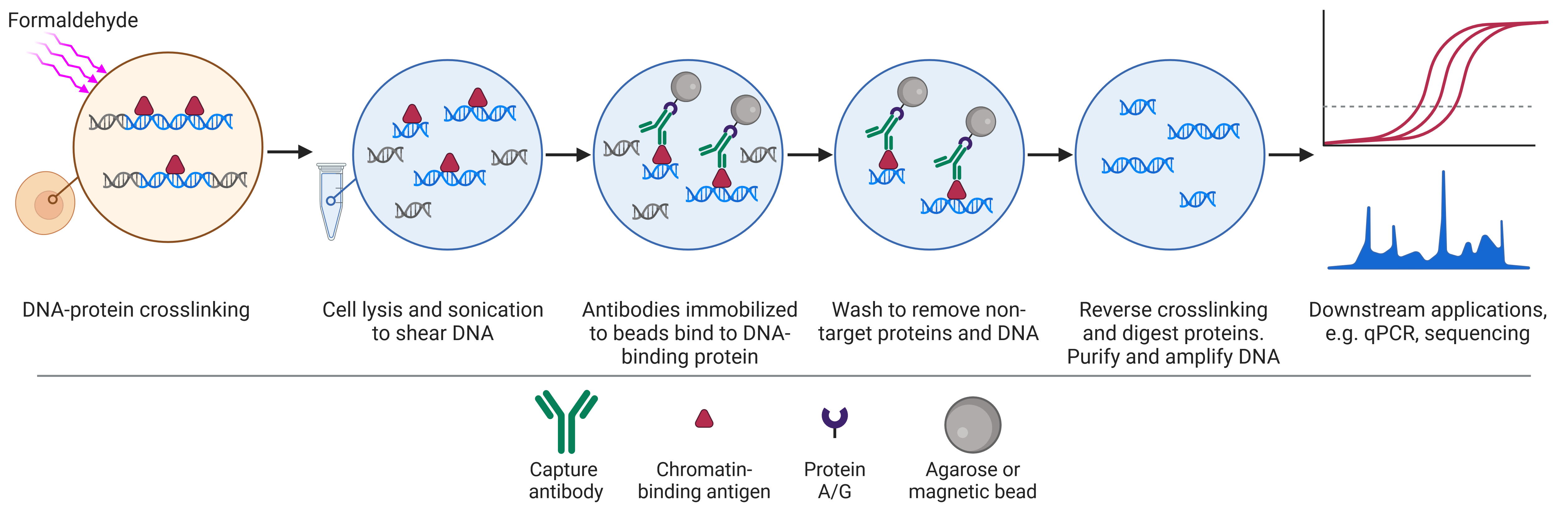
Based on the principle of immunoprecipitation (IP), ChIP relies on specific antibodies that are immobilized on a solid substrate to purify DNA-binding proteins and associated DNA. ChIP is the standard tool to study histones and the genomic regions that are associated with specific histone modifications, which regulate chromatin structure. In addition, ChIP is adept at capturing other protein-DNA interactions, such as where transcription factors bind in the genome to control gene expression.
DNA isolated by ChIP is typically investigated by quantitative PCR (qPCR) to study the expression of genes of interest, or by sequencing (ChIP-seq) for a genome-wide perspective on protein-DNA interactions.
This guide aims to provide an overview of ChIP experimental design, important controls, protocols, and troubleshooting. The critical technical aspects of ChIP will be discussed, providing an approach that can then be tailored to your specific research objectives.
Table of Contents
History of ChIP
Using antibodies to precipitate target proteins has its origins in the 1960s, when Barrett et al. referred to immunoprecipitation as a tool for measuring the concentration of gamma globulins in human serum, offering more precision than other techniques available at the time such as electrophoresis.1,2 However, ChIP did not emerge until the 1980s when it was implemented by Gilmour & Lis as a technical proof of principle, using RNA polymerase and the lac operon in bacteria, crosslinked with UV radiation.3 This was followed by the same group with investigations into the activity of RNA polymerase II and topoisomerase I at heat shock genes in untreated or heat-shocked Drosophila melanogaster cells.4–6 The use of UV in ChIP was later replaced by formaldehyde, a more efficient and reversible method of crosslinking protein to DNA.7 One of the major uses of ChIP today is to study the link between histone modifications and transcriptional activity. ChIP was first applied to this question in 1988, when Hebbes et al. found that acetylated histone H4 was enriched at genomic regions that were being actively transcribed, but not at a control locus.8
ChIP analysis initially relied on traditional molecular biology approaches such as Southern blots, later superseded by end-point PCR and qPCR.9 These techniques are useful if known genes or sequences are the subject of study, for instance to study how the association between a transcription factor and its binding sites changes with experimental treatment, but cannot reveal previously unknown regions of interaction. This limitation was overcome by ChIP-chip, in which ChIP was combined with microarray technology to provide a global view on protein-DNA interactions, first in yeast before being applied to mammalian cells in 2001.10,11 Next-generation sequencing (NGS) combined with ChIP (ChIP-seq), first performed in 2007, has since become the most popular technique for analyzing ChIP experiments, providing a rapid, high-resolution snapshot of protein-DNA interactions.12
Chromatin, Epigenetics, and Targets of ChIP
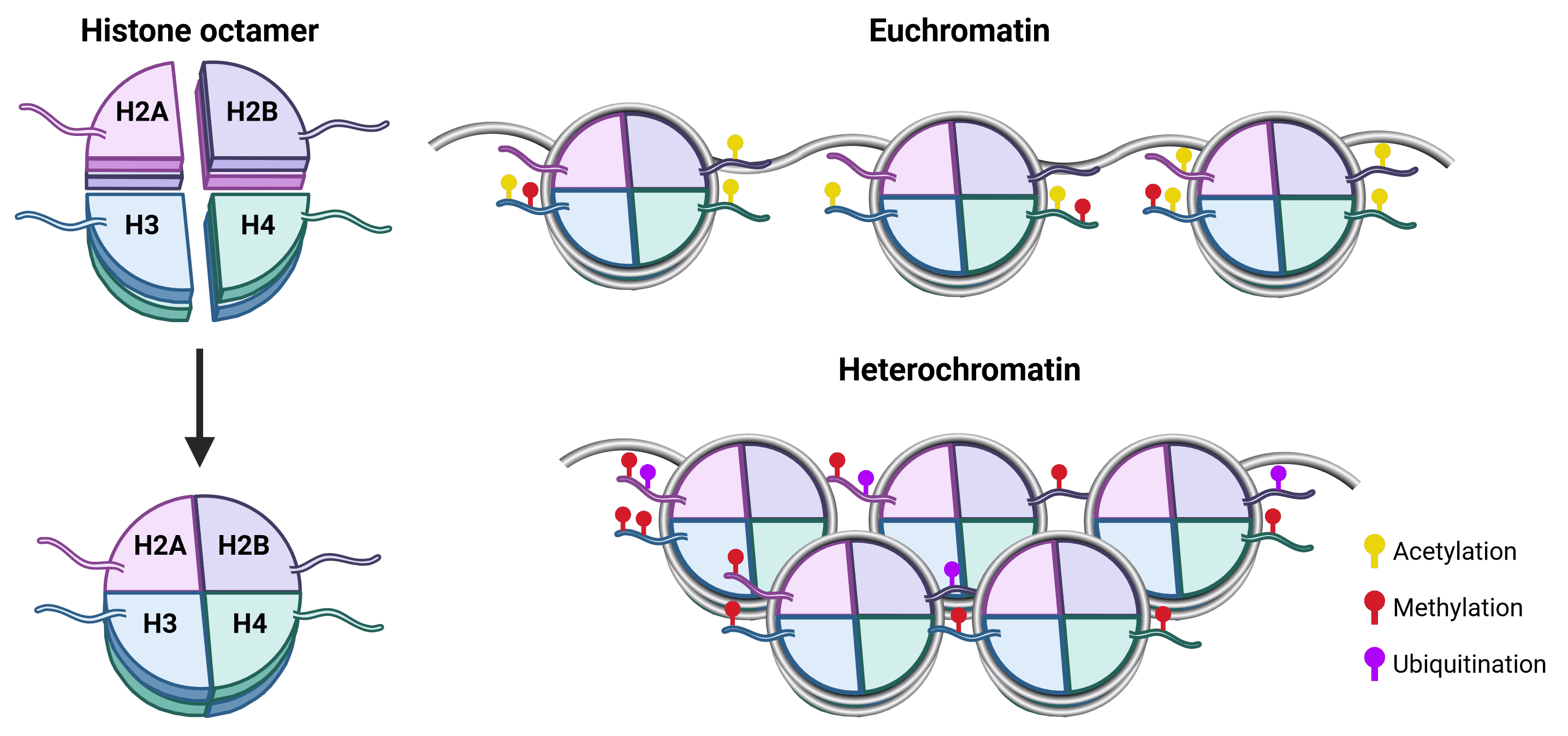
The spatial organization of genomic material is essential for appropriate gene expression.13 In eukaryotes, DNA in the nucleus is complexed with histone proteins to form chromatin, which is essential for preventing DNA strands from becoming tangled and to enable DNA packaging. The primary unit of chromatin is the nucleosome, which consists of a core of 8 histone proteins (2 copies each of H2A, H2B, H3 and H4) around which 147 bp of DNA wraps approximately 1.75 times (Figure 1). The histone H1 protein is typically referred to as a linker and has numerous functions in chromatin including the stabilization of DNA wrapping around the nucleosome and assembly of higher order chromatin structures.14

Figure 1: Nucleosome structure and chromatin organization.The histone octamer is at the core of nucleosome structure, comprising an H3-H4 tetramer and two H2A-H2B dimers around which DNA wraps.15 Each histone protein has an unstructured N-terminal (as well as an unstructured C-terminal for H2A) that protrudes from the nucleosome core and is called a tail. Histone tails are accessible to histone modifying proteins and are the prime site for PTMs, which influence whether chromatin is structured as euchromatin (transcriptionally active) or heterochromatin (transcriptionally repressed). Acetylation is generally a marker for euchromatin, because it is negatively charged and so weakens the strength of attraction between the positively charged histones and DNA.16 Methylation has no effect on charge and can be associated with either transcriptional activation or repression depending on the site.16 For example, H3K4 methylation results in gene activation, whereas H3K27 methylation leads to gene silencing. Though PTMs of H3 and H4 are the best characterized, H2A and H2B PTMs also affect chromatin structure, including methylation and ubiquitination.17
Chromatin exists in distinct, spatially segregated states that influence how accessible it is to transcriptional machinery. Heterochromatin is a tightly packed form of chromatin that is essential for genomic stability and associated with repressed transcription.18 In contrast, euchromatin is less compact and more permissive of gene expression.19
Post-translational modifications (PTMs) of histones are essential in determining chromatin state. These PTMs include phosphorylation, methylation, acetylation, ubiquitination, sumoylation, ADP ribosylation and more, which are conferred upon the histones by specific enzymes termed readers, writers and erasers.20 The combination of PTMs, particularly on histone lysine residues, are strongly associated with the dynamic regulation of gene expression by altering chromatin structure and recruiting remodeling enzymes.21
Histone PTMs are often referred to as epigenetic marks, heritable alterations to chromatin that regulate gene expression but do not change the underlying base sequence (though the exact definition has been debated).18 Other epigenetic marks include DNA methylation, chromatin remodeling, and non-coding RNA signaling.22 Unlike the base genome, epigenetic marks are dynamic, adapting to environmental cues and, along with transcription factor activity, control gene expression to determine cell fate and function.
ChIP has been used to understand the DNA binding properties of chromatin remodelers, histone-modifying enzymes, transcription factors and more. A successful ChIP experiment should take into account the nature of the ChIP target to determine the optimum approach. These include expression properties, such as how abundant the target protein is, the conditions under which it is expressed, and whether it is localized to the nucleus, and DNA-binding properties, such as if it binds to DNA directly or via another protein, how strong the association is, and how many binding loci are under investigation.
Applications and Limitations of ChIP
ChIP is a powerful tool for understanding how proteins interact with chromatin in its native environment. Fundamentally, ChIP is useful for determining which DNA sequences are being bound by the target protein, such as a transcription factor. This could identify new transcription factor binding sites, suggesting an interaction with a gene previously not known to be regulated by that protein. From this, how DNA-protein interactions change with time, treatment, cellular activity and disease state can be investigated. ChIP can also be used for identification of genetic elements, either via transcription factor binding or inferring element function based on histone modifications at the site.25
With regards to histone-DNA interactions, ChIP can identify higher order chromatin structures, such as sites of heterochromatin by trimethylation of H3 lysine 9 (H3K9me3), suggest activity or inactivity at genomic regions based on well-known histone modifications, and help determine the function of unexplored histone modifications.
Despite the discoveries enabled by ChIP, the technique has significant limitations. ChIP is a complex, multistep technique that has high cell input requirements and is slow to perform compared to comparable techniques such as CUT&RUN.23 ChIP can yield low signal and have high background, which is particularly true if an antibody of insufficient specificity is used, such as one that cannot adequately distinguish between mono-, di- or tri-methylation of a histone. The resolution of ChIP is limited to the size of DNA fragment that is precipitated, typically ~150-500 bp, meaning precise binding sites cannot be determined without other techniques. Further, the function of a protein at a given site cannot be inferred from ChIP alone, such as if it is activating or repressing transcription. With X-ChIP, the protein may not be directly interacting with the DNA, the epitope of the protein could be masked by cofactors, while crosslinking can fix irrelevant proteins that only transiently interact with the DNA but have no functional significance.
Sample Preparation
Sample type
ChIP can be performed on both cells and tissue, though different cell types in a tissue will have heterogeneous chromatin structure and histone marks, which will confound data interpretation. Tissue can be either fresh or frozen, and approximately 1-5 mg of tissue is required for adequate material,26 though this will vary with cell type, target and application.
Cell Number
An average of 4x106 cells is recommended as a starting point for a ChIP procedure, but this is dependent on the abundance of the protein target and how many individual proteins are present at each locus. For instance, fewer cells are needed if analyzing RNA polymerase or histones compared to transcription cofactors. The downstream application also influences how much DNA is required for further analysis, with qPCR requiring very little (in the picogram range, 1x105 cells) and ChIP-seq requiring more (1-10 ng, 1x106 cells).
Crosslinking Proteins to DNA
Crosslinking stabilizes and preserves protein-DNA interactions and is necessary when performing ChIP on any non-histone proteins. Histones can be precipitated without crosslinking in N-ChIP, although new modifications to the histones may occur in the absence of crosslinking if enzymes are still active.
Crosslinking in X-ChIP can be performed with UV light, formaldehyde, or other chemical crosslinkers, but formaldehyde is the standard approach because it is a reversible and highly efficient crosslinking agent. With a crosslinking distance of only 2 Å (0.2 nm), crosslinking by formaldehyde results in the preservation only of proteins closely associated with DNA, though it will also fix proteins to proteins, so cofactors can be co-precipitated to facilitate the study of indirect protein-DNA interactions. For more distal interactions or larger protein complexes, other crosslinkers such as DSG (7.7 Å) or EGS (16.1 Å) can be used instead.
Formaldehyde is typically used at 1% concentration at room temperature for 10 minutes, but a performing a time course pilot experiment is recommended using a range of 2-30 minutes to determine the optimal crosslinking time. Under crosslinking may result in dissociation of the protein-DNA complexes during the ChIP procedure, while crosslinking for too long can mask epitope sites, inhibit chromatin fragmentation, and making reversal of crosslinking more difficult. At the end, glycine (~150 mM final concentration) can be added to quench the crosslinking.
Cell Lysis
Cell lysis is necessary for the release of chromatin from the cell. While whole cell lysis can be performed in one step, isolating nuclei by first removing the cytoplasm tends to result in lower background. As with IP, lysis in ChIP is achieved using buffers containing a mixture of detergents and salts, with the primary aims being to stabilize proteins, inhibit enzyme activity, and rupture membranes to release cellular components.
The cell membrane can be more readily disrupted than the nuclear membrane, meaning weaker, non-ionic detergents such as NP-40 or Triton X-100 can be used to isolate nuclei. This can then be followed by a nuclear lysis step using both mechanical force (e.g. dounce homogenizer, vortexer, or passing the solution through a needle) and a buffer that contains stronger detergents such as sodium dodecyl sulfate (SDS). The inclusion of the crosslinking step in ChIP makes protein-DNA interactions less susceptible to being disrupted by detergents than protein-protein interactions are in co-IP.
In addition to detergents, ChIP lysis buffers tend to contain NaCl and/or Tris-HCl, and usually have a slightly basic pH (7.4 to 8), though this can be optimized (between pH 6-9). Other buffer components that can be optimized include: salts (0-1 M) for maintaining ionic strength and correct tonicity for easy cell lysis; divalent cations such as Mg2+ (0-10 mM) which can help to prevent DNA causing the solution to become viscous; EDTA (0-5 mM) for chelating ions such as Zn2+ that proteases require for proteolytic function; and enzyme inhibitors, discussed further below.
Successful lysis can easily be checked under a microscope by comparing 10 µl of sample before and after lysis on a hemocytometer, which will confirm the release of nuclei and subsequent nuclear lysis. Cell lysis varies depending on the cell type, so should be optimized by gradually increasing the stringency of buffer components, increasing incubation time, or by using mechanical force. Regardless of the composition of the buffer, performing all steps of a ChIP at 4°C or on ice is strongly recommended in order to minimize sample degradation.
Enzyme Inhibitors
The final components of lysis buffers are inhibitors of proteases and enzymes that alter protein PTMs, particularly phosphatases. Protease inhibitors prevent the degradation of target proteins, while PTMs are often necessary for protein-protein interactions and so must be maintained for co-IP. Preserving PTMs is obviously essential if the target of the IP is to understand the PTM state of target proteins.
Inhibitors should be added fresh, immediately before lysis buffer use. Table 2 and Table 3 contain common inhibitors used in lysis buffers.
| Protease inhibitor | Typical concentration | Solvent | Target |
| Aprotonin | 1-10 µg/ml | Water | Serine proteases: trypsin, chymotrypsin, kallikrein, plasmin |
| Benzamidine | 15 µg/ml | Water | Serine proteases: trypsin, plasmin, thrombin, factor Xa |
| EDTA/EGTA | 1-10 mM | Water | Metallo proteases |
| Leupeptin | 1-2 µg/ml | Water | Serine and cysteine proteases: plasmin, trypsin, papain, cathepsin B, thrombin, calpain |
| PMSF | 0.1-1 mM | Methnaol | Serine and cysteine proteases: trypsin, chymotrypsin, thrombin, papain |
| Pepstatin A | 1-3.5 µg/ml | Methanol | Aspartic acid proteases: pepsin, cathepsin D, renin, chymosin, HIV- and MMTV-proteases |
Table 2: Commonly used protease inhibitors in co-IP. Note that protease inhibitor cocktails are commercially available (often as tablets) and provide a convenient way to ensure protease inhibition.
| Phosphatase inhibitor | Typical concentration | Solvent | Target |
| β-Glycerophosphate | 1 mM | Water | Serine and threonine phosphatases |
| Okadaic acid | 10-1000 nM | DMSO or ethanol | Protein phosphatase 1/2a |
| Sodium fluoride | 10 mM | Water | Serine and threonine phosphatases |
| Sodium orthovandate | 1 mM | Water | Tyrosine phosphatases |
Table 3: Commonly used phosphatase Inhibitors in co-IP. Phosphatase inhibitor cocktails are commercially available (often as tablets) and provide a convenient way to ensure phosphatase inhibition. Note that while phosphatase inhibitors are typically included in IP buffers, inhibitors of other PTMs, such as ubiquitination and methylation, are included only as needed, determined by the aims of the experiment.
Chromatin Fragmentation
Chromatin in its native form is high molecular weight, and so must be fragmented to render the chromatin soluble and accessible to the ChIP antibody. Chromatin can be fragmented either by sonication or by enzymatic digestion, with sonication being more common. Shearing by sonication should only be used with X-ChIP, because sonication will disrupt protein-DNA interactions without crosslinking in N-ChIP. Table 4 below shows a comparison between the two approaches.
| Sonication | Enzymatic |
| Application | X-ChIP only | X-ChIP and N-ChIP |
| Tool used | Probe or bath sonicator | Micrococcal nuclease (MNase) |
| Fragment site | Random | Preferentially cuts at AT-rich sequences and internucleosome regions. Difficulty cutting in heterochromatin regions |
| Hands-on time | Labor-intensive, especially with probe sonicator | More samples can be processed at once |
| Reproducibility | More variable between runs – ensure probe of probe sonicator does not touch the side of the tube, is at the same depth in the sample, and is cleaned between samples. Water bath sonicators can have hotspots, resulting in non-uniformity | Highly reproducible, but enzymatic activity may vary over time or between lots |
| Impact on samples | Generates heat and can cause foaming of sample, which can reduce chromatin and protein integrity and cause dissociation. Sonication should be pulsed and performed on ice to reduce heat | Much milder. Nuclei must be isolated first |
| Optimization | Number of cycles. Pulse duration and pulse intensity can be increased as well, but may increase heat generated | Enzyme concentration, incubation time |
Table 4: Comparison of major features of X-ChIP and N-ChIP.
Regardless of which approach is taken, chromatin fragmentation must be optimized for each cell type to give fragments of the appropriate length. The ideal fragment length is generally thought to be 200-750 bp, though some studies have found that 150-300 bp (1 to 2 nucleosome units) with an average of 200-250 bp was optimal for ChIP-seq.27,28 Some of the sample can be examined by agarose gel electrophoresis to determine the optimal fragmentation conditions (Figure 3), and ideally would be performed during each experiment to confirm ideal fragment length. Prior to gel electrophoresis, DNA samples should be cleaned up by reversing the crosslinking (e.g. by heating at 65°C) and treating with proteinase K and RNase A to remove protein and RNA, respectively, that might otherwise interfere. This is then followed by DNA purification using a spin column or phenol-chloroform extraction.
Figure 3: Optimizing DNA fragment length. Schematic example of DNA run on an agarose gel after sonication in which sonication parameters (pulse width and power) and cell number are kept consistent, but samples are sonicated for different lengths of time. In this instance, the optimal time would be 10 or 15 minutes, depending on downstream application.
Antibody Selection
Antibodies are generated by the immune system of a host organism (e.g. mouse, rabbit) that has been repeatedly immunized with a specific antigen. The antibodies recognize and specifically bind to that antigen with a high affinity, allowing the protein to be precipitated from the lysate. High specificity is essential for a successful ChIP, as non-specific binding can result in high background or false positive results, and ChIP targets often only differ by small chemical groups such as PTMs. For example, mono-, di and tri-methyl groups on histones can indicate different activity states: H3K9me1 is generally an activating mark, but H3K9me2 is generally a repressive mark.
To confirm the specificity of the antibody-antigen interaction, an isotype control should be included. This control is often called a “mock” IP, and consists of a non-immune antibody of the same isotype as the experimental antibody. This should not have any affinity for the target antigen, but it may non-specifically bind to other factors. Therefore, any DNA sequences that emerge from both the ChIP and the isotype control conditions are unlikely to be specifically bound by the protein of interest.
The following factors should be taken into account when selecting an antibody.
Reactivity
Primary antibodies will recognize antigens only from certain species, which will be the species that the immunizing antigen was originally from, as well as closely related species. For example, an antibody that recognizes a mouse protein target will often also recognize the same protein in rat, but possibly not in fish. The key determinant of this is how similar the epitope sequence is between species, allowing researchers to predict if an available antibody will work in a non-validated species.
Concentration
Antibody concentration should be carefully optimized. Too little antibody will result in less protein being precipitated, and therefore less DNA to take forward to qPCR or sequencing. Conversely, too much antibody can increase background and non-specific binding. Determine antibody concentration empirically, with a starting point of 0.5-2 μg of antibody used per 10 μg of chromatin DNA (approximately 4 x 106 cells).
Clonality
Both monoclonal and polyclonal primary antibodies can be used to detect the protein of interest in ChIPs. Monoclonal antibodies correspond to a single epitope for a given antigen, whereas polyclonal antibodies may recognize multiple epitopes. As a result, polyclonal or oligoclonal (mixtures of monoclonal) antibodies are often preferred to monoclonals for ChIP because they offer a high chance of capturing the target, and a smaller chance that the interaction site between bait and prey will be blocked.
Application Compatibility
Co-IP, ChIP and RIP always seek to isolate proteins in their native conformation, which is essential for physiologically relevant molecular interactions, so a chosen antibody should recognize this form rather than denatured protein (as is common in western blotting). This contrasts to individual protein IPs, which can be performed on denatured samples. If an IP-validated antibody is not available, it is usually acceptable to use one that has been validated for IHC or ELISA, which also work with native proteins. To confirm that a protein target has been precipitated before going on to PCR or sequencing, perform a western blot by boiling the bead-protein-DNA complexes in loading buffer after the last wash and running SDS-PAGE. While this is useful for confirming that an antibody is working, it does not necessarily mean that the target protein will be crosslinked to DNA, particularly for indirect interactions.
Bead support
The type of beads chosen when performing ChIP is an important consideration and will dictate certain aspects of the overall protocol. Beads are usually either agarose (or Sepharose, which is a tradename for a crosslinked, beaded-form of agarose) or magnetic beads. Table 5 summarizes the key differences between agarose and magnetic beads, which are described further below.
| Agarose | Magnetic |
| Size | 50-150 μm | 1-4 μm |
| Uniformity | Low | High |
| Binding capacity | High | Medium |
| Diffusion | Slow | Fast |
| Washing | Extensive washing Easier to pick up some of the pellet when removing supernatant | Washing steps reduced Less chance of proteolytic damage |
| Yield | Medium | High |
| Time required | 60-90 minutes | 30-60 minutes |
| Automation | No | Yes |
| Reproducibility | Medium | High |
Table 5: Key features of agarose and magnetic beads in ChIP.
Agarose Beads
Agarose beads tend to be 50-150 μm in size, and they have a very high binding capacity because they are porous, creating a very high surface area per bead that is available for binding to antibodies. However, this can create a requirement to use a large amount of antibody to ensure that every surface of the agarose beads is covered in antibody, lest any unoccupied agarose non-specifically bind to lysate components and increase background signal. To solve this issue, it is possible to back-calculate from the amount of expected analyte to the amount of antibody needed for detection, to the amount of agarose needed to hold the antibody. The downside is that this approach requires significant knowledge or prior experience to estimate such quantities. An alternative would be to simply saturate the beads with excess antibody, but antibody is typically expensive and limiting. The best option is therefore to pre-clear the lysate to remove anything that will non-specifically bind to the agarose.
Magnetic Beads
Magnetic beads lack the porosity of agarose because they are solid spheres, resulting in only the external surface being available for binding. While this may at first glance suggest that magnetic beads have much lower binding capacity, they are much smaller than agarose particles at 1-4 μm, meaning that there can be far more beads per unit volume, so the ultimate binding capacities of the two are similar, with agarose maintaining a small advantage.
Magnetic beads offer several other advantages over agarose for use in ChIP, however. They are highly uniform in size and have a consistent binding capacity, which can make experiments more consistent between runs. Magnetic beads are manipulated using powerful magnets that pull the beads to one side of a tube so that the supernatant can be removed, obviating the need for centrifugation. Centrifugation can be stressful for complexes, which may be a consideration for N-ChIP, but crosslinking should preserve interactions in X-ChIP. Magnet-based manipulation is also faster and makes ChIPs more amenable to higher throughput applications.
Immobilizing Antibodies to Beads
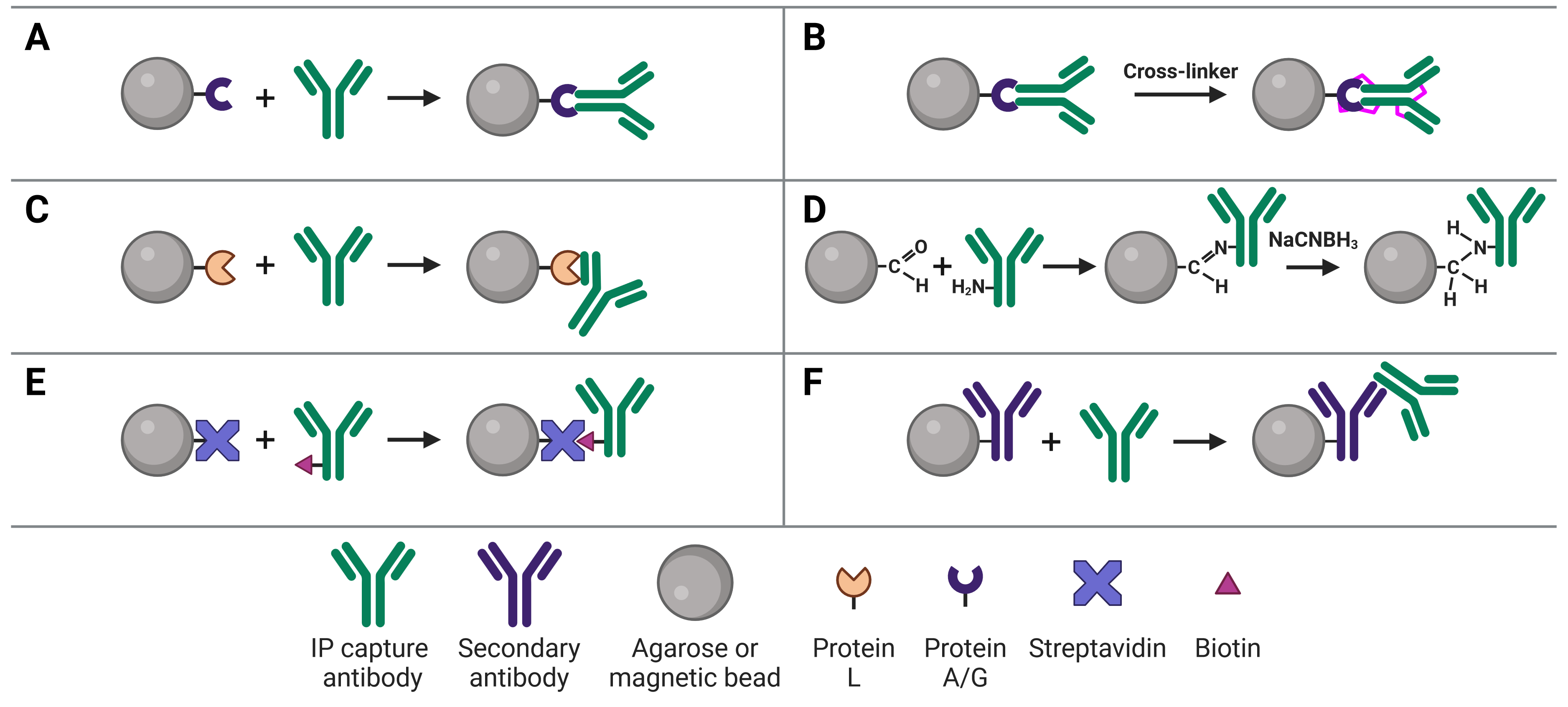
Immobilizing antibodies to the solid phase (beads) is essential for ChIP, enabling the precipitation of the target protein-DNA complexes. While the most common method is to use Protein A or G to capture the ChIP antibody, various approaches can be used, each of which has its own benefits and applications. These approaches are illustrated in Figure 4 and explained in more detail below.
Figure 4: Approaches to IP antibody immobilization on beads.A, Protein A, G or A/G bind to the Fc region of antibodies. B, Antibodies can be crosslinked to Protein A, G or A/G to prevent eluting the antibodies during antigen elution. C, Protein L binds to the light chain of antibodies. D, Direct immobilization of antibodies to beads obviates the need for Protein A, G or L, and prevents antibody elution. E, Streptavidin-conjugated beads capture biotinylated IP antibodies with very high affinity. F, Secondary antibodies directly bound to beads can be used to capture IP antibodies from a specific host or of a specific isotype, but offer less flexibility than Protein A or G.
Protein A and Protein G
The most common approach to immobilize the antibody to the chosen bead is with Protein A29 or Protein G,30 which are covalently bound to the beads following bead activation with a coupling agent such as cyanogen bromide or N-hydroxysuccinimide (NHS). Protein A and Protein G are derived from bacteria, and bind to the heavy chains of the antibody’s Fc region (Figure 4a). Binding to this site has the advantage of orienting the antibody such that the Fab region is clear and directed away from the bead to bind to the target protein.
The choice between Protein A and G will depend on the capture antibody’s host species and isotype, because Protein A and G exhibit different binding affinities for different immunoglobulins (Table 6). To circumvent this issue, the recombinant Protein A/G was engineered to contain four Protein A and two Protein G binding sites, and which binds all of the subtypes that Protein A and G bind individually.
| Species | Ig subclass | Protein A binding | Protein G binding |
| Human | IgA | ++ | X |
| IgD | ++ | X |
| IgE | ++ | X |
| IgG1 | ++++ | ++++ |
| IgG2 | ++++ | ++++ |
| IgG3 | X | ++++ |
| IgG4 | ++++ | ++++ |
| IgM | ++ | X |
| Mouse | IgG1 | + | ++ |
| IgG2a | ++++ | ++++ |
| IgG2b | +++ | +++ |
| IgG3 | ++ | +++ |
| IgM | +/X | + |
| Rat | IgG1 | X | + |
| IgG2a | X | ++++ |
| IgG2b | X | ++ |
| IgG2c | + | ++ |
| IgM | +/X | X |
| Chicken | IgY | X | + |
| Cow | IgG | ++ | ++++ |
| Goat | IgG | +/X | ++ |
| Guinea pig | IgG | ++++ | ++ |
| Hamster | IgG | + | ++ |
| Pig | IgG | +++ | +++ |
| Rabbit | IgG | ++++ | +++ |
| Sheep | IgG | +/X | ++ |
Table 6: Binding affinities of Protein A and Protein G for different immunoglobulins. ++++ refers to strong binding, + is weak binding, X is no binding affinity.
The interaction between antibodies and Protein A/G will occur in most physiological buffers, but binding, and therefore capacity, can be increased by using dedicated Protein A or Protein G binding buffers. Protein A binds IgG best at pH 8.2, whereas protein G binds best at pH 5. However, these buffers may then not be suited for antigen binding, thus optimization of binding buffers is always necessary.
Covalently Linking Capture Antibodies to Protein A/G
Binding of a capture antibody to the solid phase via Protein A/G is not covalent, meaning the antibody will be eluted along with the target antigen at the end of the experiment. However, unlike with IP and co-IP, ChIP samples are digested with proteinase K at the end of the procedure to isolate the DNA, so this is rarely an issue.
If it does cause an issue, such as if the protein content of the ChIP is needed for downstream assays as well, the ChIP antibody can be covalently linked to Protein A/G (Figure 4b). Covalent linkage is achieved using a cross-linker such as disuccinimidyl suberate (DSS) or bissulfosuccinimidyl suberate (BS3). These are simple carbon spacers with NHS ester groups at either end, which react with primary amines on lysine residues in the proteins to form a covalent link between them.
Protein L
Protein L is derived from Peptostreptococcus magnus and binds to the kappa light chain in the variable domain of antibodies (Figure 4c).31 Protein L is most commonly used with mouse and rat IgM capture antibodies, which bind poorly to Protein A/G. This is due to the heavy chains of IgMs interacting with each other, forming their classic pentameric structure and blocking Protein A/G binding.
Direct Immobilization
Direct immobilization refers to covalent bonding of the antibody to the beads without Protein A, G or L acting as an intermediary (Figure 4d). This is achieved by activating beads with aldehyde groups, which are highly reactive with primary amine groups on antibodies, followed by reduction with sodium cyanoborohydride to form a stable secondary amine bond. This approach can be useful if Protein A/G/L are not compatible with the subclass of ChIP antibody. Removing Protein A/G as a component in the system also removes it as a source of non-specific binding during the ChIP.
The slight disadvantage of this approach is that antibodies are coupled to the bead in a random orientation, as opposed to the directed orientation that results from Protein A/G binding. However, this only has a minor effect on capacity and ChIP yield.
Other Immobilization Methods
Biotin-avidin binding
Avidin is a protein found in egg whites, while streptavidin is from purified from the bacterium Streptomyces avidinii, but they both have an extremely high and specific affinity for biotin. Biotin is a small 244 Da vitamin that is easy to covalently bond to protein (the carboxyl group in biotin can be modified with reactive groups such as NHS esters, maleimides or hydrazides that target amines (-NH2), sulfhydryls (-SH) or aldehydes (>C=O), respectively, on proteins).
The affinity between biotin and avidin is extremely strong and specific, making it an ideal system for affinity purification, in which a biotinylated antibody is bound by streptavidin-conjugated beads (Figure 4e). Only harsh buffers can dissociate biotin-avidin, such as 8 M guanidine-HCl at pH 1.5, meaning more stringent washes can be applied to reduce background.
Secondary antibodies
An indirect ChIP method that is analogous to indirect methods used in IHC, ELISA and WBs uses secondary antibodies directly immobilized to beads (Figure 4f). These secondary antibodies recognize the host species of the ChIP capture antibody to ensure specificity. For example, a mouse ChIP antibody could be bound by a goat anti-mouse secondary antibody attached to the beads. The disadvantage of this approach is that it does not offer the flexibility of Protein A/G-conjugated beads, which will bind to a ChIP antibody from any host.
Pre-clearing and Pre-blocking
Pre-clearing is an optional but often worthwhile step to reduce non-specific binding in ChIP. Pre-clearing refers to incubating samples with the beads (including Protein A/G or other attachment substrates) in order to remove lysate components that bind to bead non-specifically. This prevents these components from being carried through and ultimately eluted, therefore reducing background signal.
Pre-clearing is not necessary if the target is particularly abundant, and is less important if using magnetic beads over agarose beads, because magnetic beads are less prone to non-specific binding.
Prior to pre-clearing, 1-10% of the chromatin should be conserved before the addition of any antibody or beads. This represents the starting material and will be run alongside samples during qPCR or sequencing analysis to normalize ChIP-enriched sequences.
Reducing non-specific binding can also be achieved by pre-blocking the bead. This works in a similar way to blocking in immunostaining, western blotting and ELISA. The bead is incubated with a mild blocking buffer containing 0.1-1% BSA, non-fat milk or 0.1-1% Tween-20 to block sites of non-specific binding on the bead, preventing lysate components from binding. Blocking non-specific DNA binding to beads can be achieved by blocking with salmon sperm DNA, though this is not recommended for ChIP-seq applications because of contaminating reads.
Immunoprecipitation
As in IP and co-IP, there are two approaches for ChIP: the pre-immobilized antibody method in which the antibody is incubated with the bead first, and the free-antibody method in which the antibody is added to the chromatin first before the beads are added. Using free antibodies can be beneficial in instances where the target protein is low abundance as it can give the highest yield, though it can also give higher background than the pre-immobilized approach. The pre-immobilized method must be used when using direct immobilization approaches. Once the approach has been decided upon, the antibodies are added to the sample and incubated overnight at 4°C.
Washing
After the antibodies and beads have been added, the bead-antibody-antigen complexes are pelleted, allowing the supernatant to be removed. The beads then need to be washed to remove non-specific binding of other lysate components to the bead, immobilization substrate or antibody. Washing is usually done using multiple dedicated washing buffers of increasing stringencies, including low salt (NaCl), high salt, and lithium chloride (LiCl) buffers, before a final Tris-EDTA (TE) buffer wash to prepare the DNA for elution.
Other components of the wash buffers include Tris-HCl as a base, containing detergents such as 0.1% SDS and 0.5-1% NP-40 or Triton X-100 to reduce non-specific binding. Salt (NaCl) can be increased up to 1 M to increase the stringency of the wash by reducing ionic and electrostatic interactions, while LiCl provides even higher stringency. These buffers will also contain EDTA to inhibit DNases, as well as protease and phosphatase inhibitors.
Elution and Crosslink Reversal
Elution refers to dissociating the target protein-DNA complex from the beads to obtain DNA for analysis. Elution buffers usually contain sodium carbonate and SDS.
To obtain a pure DNA sample, however, the formaldehyde crosslinks must be reversed and contaminants removed. Heating at 95°C for 15 minutes may be sufficient to reverse crosslinks, but sometimes long heat treatment at 65°C alongside proteinase K treatment is recommended. RNase can also be added to remove contaminating RNA. Finally, the DNA sample should be cleaned up using a phenol-chloroform extraction or with spin columns.
Analysis
The typical aim of ChIP experiments is to identify specific genomic regions that are occupied by a given DNA-binding protein or modified histone protein. qPCR and NGS are the two most common ways of assessing this.
ChIP-qPCR
qPCR is used in conjunction with ChIP to assess the relative occupancy of a DNA-binding protein at a few known or hypothesized DNA sequences. qPCR allows researchers to quantify the enrichment of a given sequence over the amount of that sequence in either the input control or the mock (isotype control) ChIP. It is also useful to confirm that a ChIP has worked by assessing known regions by qPCR before performing full sequencing on samples.
In qPCR, polymerases amplify sequences within the DNA sample using specific primer pairs. High efficiency (95-105%) primer pairs are important for qPCR, because otherwise off-target sequences will be amplified. Unlike end-point PCR, qPCR provides a real-time read-out via fluorescent reporter of how much of a given sequence is present within the sample, allowing differences between samples (or samples and controls) to be precisely quantified.
Fluorescent reporters come in the form of either TaqMan or SYBR Green. TaqMan requires probes that recognize and bind to specific sequences and that contain a fluorescent reporter and a quencher. While bound, the quencher prevents fluorescent signal, but during amplification the endogenous 5' nuclease activity of the polymerase cleaves the probe, separating the dye from the quencher and allowing fluorescent signal. In contrast, SYBR green does not require any specific probes, and instead fluoresces when it intercalates into double stranded DNA. The more dsDNA (as amplification progresses), the greater the signal, but this approach is therefore more susceptible to non-specific amplification and primer-dimer.
The fluorescent signal from qPCR can be used to indicate the cycle threshold (Ct) for a given sample, which is the point at which the fluorescent signal exceeds background. The amount of DNA in the sample directly corresponds to the Ct, therefore Ct values can be used to calculate how much of a given sequence is in a sample and compare groups in relative or absolute terms.
ChIP-seq
NGS allows researchers to take an unbiased view of all of the sites where a DNA-binding protein or histone modification is associated with in the genome. These sites are often referred to as “peaks”, with sharp peaks corresponding to a high resolution sequence determination of where a protein is binding.
While ChIP-seq provides far more information than qPCR, it also requires more sample processing time to ligate adaptors to samples during NGS library preparation, and bioinformatics expertise for analysis. Sequencing also adds a significant cost to a ChIP experiment, although NGS has become significantly cheaper than when ChIP was first developed, making it more accessible.
References
Diagrams created with BioRender.com.
- Barrett, B., Wood, P. A. & Volwiler, W. Quantitation of gamma globulins in human serum by immunoprecipitation. J. Lab. Clin. Med. 55, 605–615 (1960).
- DeCaprio, J. & Kohl, T. O. Immunoprecipitation. Cold Spring Harb. Protoc. 2020, pdb.top098509 (2020).
- Gilmour, D. S. & Lis, J. T. Detecting protein-DNA interactions in vivo: distribution of RNA polymerase on specific bacterial genes. Proc. Natl. Acad. Sci. U. S. A. 81, 4275–4279 (1984).
- Gilmour, D. S. & Lis, J. T. In vivo interactions of RNA polymerase II with genes of Drosophila melanogaster. Mol. Cell. Biol. 5, 2009 (1985).
- Gilmour, D. S. & Lis, J. T. RNA polymerase II interacts with the promoter region of the noninduced hsp70 gene in Drosophila melanogaster cells. Mol. Cell. Biol. 6, 3984–3989 (1986).
- Gilmour, D. S., Pflugfelder, G., Wang, J. C. & Lis, J. T. Topoisomerase I interacts with transcribed regions in Drosophila cells. Cell 44, 401–407 (1986).
- Solomon, M. J., Larsen, P. L. & Varshavsky, A. Mapping protein-DNA interactions in vivo with formaldehyde: Evidence that histone H4 is retained on a highly transcribed gene. Cell 53, 937–947 (1988).
- Hebbes, T. R., Thorne, A. W. & Crane‐Robinson, C. A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J. 7, 1395–1402 (1988).
- Mundade, R., Ozer, H. G., Wei, H., Prabhu, L. & Lu, T. Role of ChIP-seq in the discovery of transcription factor binding sites, differential gene regulation mechanism, epigenetic marks and beyond. Cell Cycle 13, 2847–2852 (2014).
- Blat, Y. & Kleckner, N. Cohesins bind to preferential sites along yeast chromosome III, with differential regulation along arms versus the centric region. Cell 98, 249–259 (1999).
- Weinmann, A. S., Bartley, S. M., Zhang, T., Zhang, M. Q. & Farnham, P. J. Use of Chromatin Immunoprecipitation To Clone Novel E2F Target Promoters. Mol. Cell. Biol. 21, 6820–6832 (2001).
- Robertson, G. et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 4, 651–657 (2007).
- Gibcus, J. H. & Dekker, J. The Hierarchy of the 3D Genome. Mol. Cell 49, 773–782 (2013).
- Cutter, A. & Hayes, J. J. A Brief Review of Nucleosome Structure. FEBS Lett. 589, 2914–2922 (2015).
- Luger, K. Structure and dynamic behavior of nucleosomes. Curr. Opin. Genet. Dev. 13, 127–135 (2003).
- Singh, D., Nishi, K., Khambata, K. & Balasinor, N. H. Introduction to epigenetics: basic concepts and advancements in the field. in Epigenetics and Reproductive Health vol. 21 xxv–xliv (2020).
- Wang, H. et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature 431, 873–878 (2004).
- Allshire, R. C. & Madhani, H. D. Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 19, 229–244 (2018).
- Morrison, O. & Thakur, J. Molecular Complexes at Euchromatin, Heterochromatin and Centromeric Chromatin. Int. J. Mol. Sci. 22, 6922 (2021).
- Biswas, S. & Rao, C. M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 837, 8–24 (2018).
- Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011).
- Wu, Y.-L. et al. Epigenetic regulation in metabolic diseases: mechanisms and advances in clinical study. Signal Transduct. Target. Ther. 8, 1–27 (2023).
- Skene, P. J. & Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6, e21856 (2017).
- Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019).
- Gade, P. & Kalvakolanu, D. V. Chromatin Immunoprecipitation Assay as a Tool for Analyzing Transcription Factor Activity. in Transcriptional Regulation: Methods and Protocols, 85–104 (2012).
- Cotney, J. L. & Noonan, J. P. Chromatin Immunoprecipitation with Fixed Animal Tissues and Preparation for High-Throughput Sequencing. Cold Spring Harb. Protoc. 2015, pdb.prot084848 (2015).
- Kidder, B. L., Hu, G. & Zhao, K. ChIP-Seq: technical considerations for obtaining high-quality data. Nat. Immunol. 12, 918–922 (2011).
- Keller, C. A. et al. Effects of sheared chromatin length on ChIP-seq quality and sensitivity. G3 Genes|Genomes|Genetics 11, jkab101 (2021).
- Hjelm, H., Hjelm, K. & Sjöquist, J. Protein A from Staphylococcus aureus. Its isolation by affinity chromatography and its use as an immunosorbent for isolation of immunoglobulins. FEBS Lett. 28, 73–76 (1972).
- Björck, L. & Kronvall, G. Purification and some properties of streptococcal protein G, a novel IgG-binding reagent. J. Immunol. Baltim. Md 1950 133, 969–974 (1984).
- Nilson, B. H., Solomon, A., Björck, L. & Akerström, B. Protein L from Peptostreptococcus magnus binds to the kappa light chain variable domain. J. Biol. Chem. 267, 2234–2239 (1992).